Vikings, Vikings, Vikings! Hordes of high quality ancient DNA
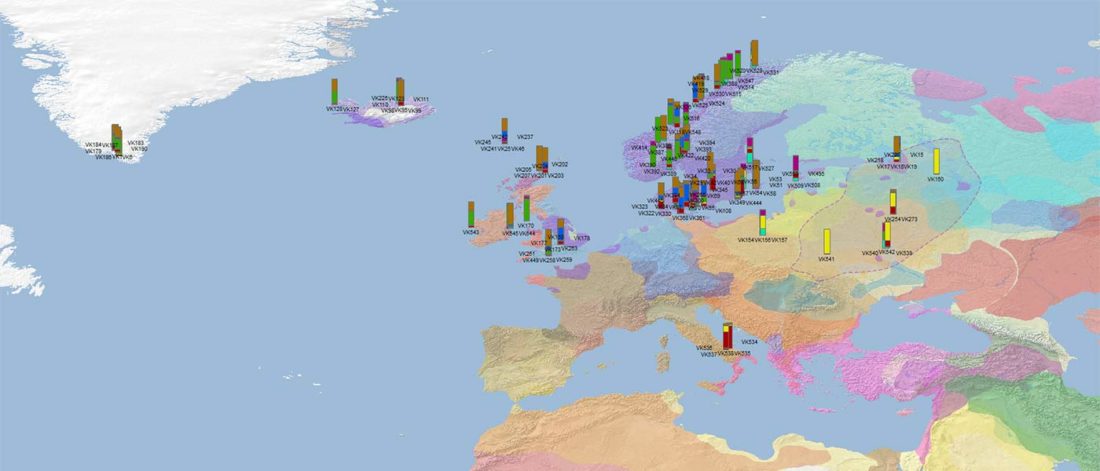
Recent paper (behind paywall) Population genomics of the Viking world, by Margaryan et al. Nature (2020), containing almost exactly the same information as its bioRxiv preprint.
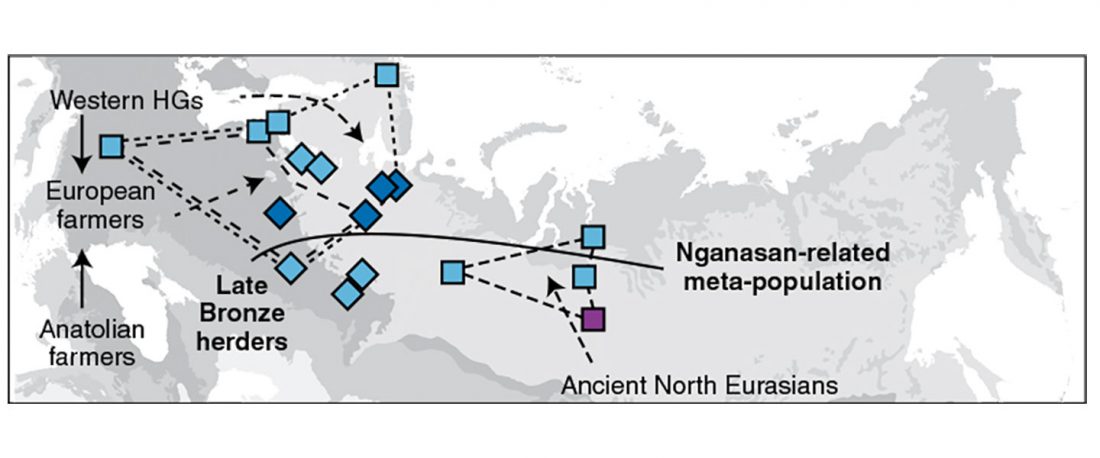
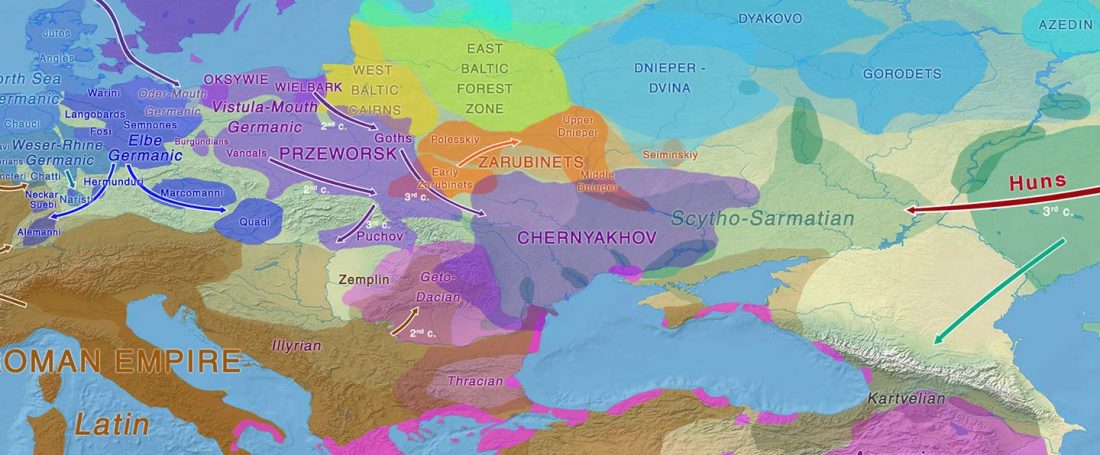
I have used Y-SNP inferences recently reported by FTDNA (see below) to update my Ancient DNA Dataset and the ArcGIS Online Map, and also to examine the chronological and geographical evolution of Y-DNA (alone and in combination with ancestry).
Sections of this post:
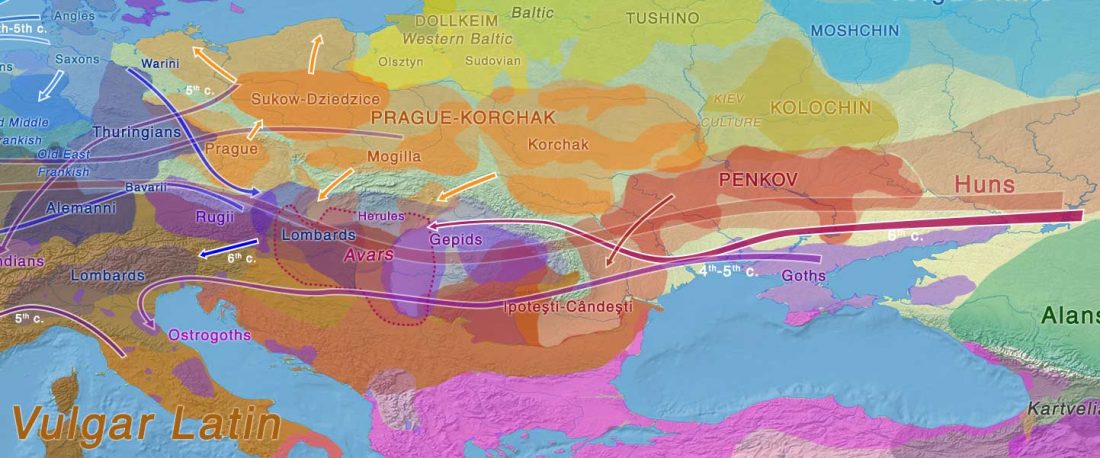
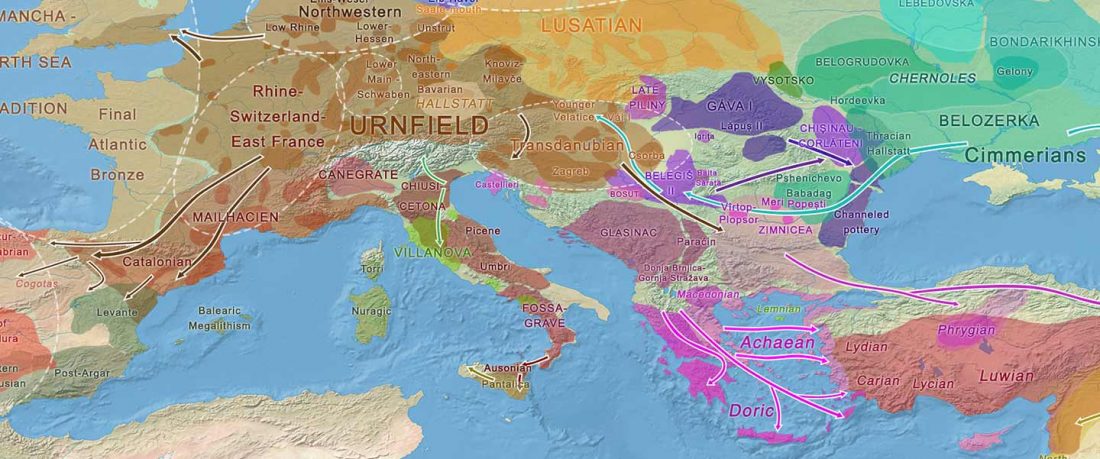
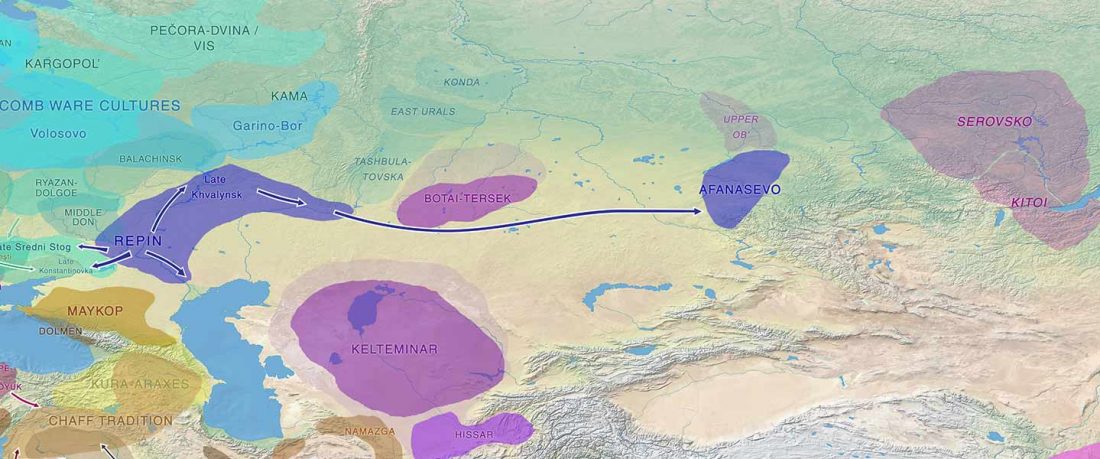
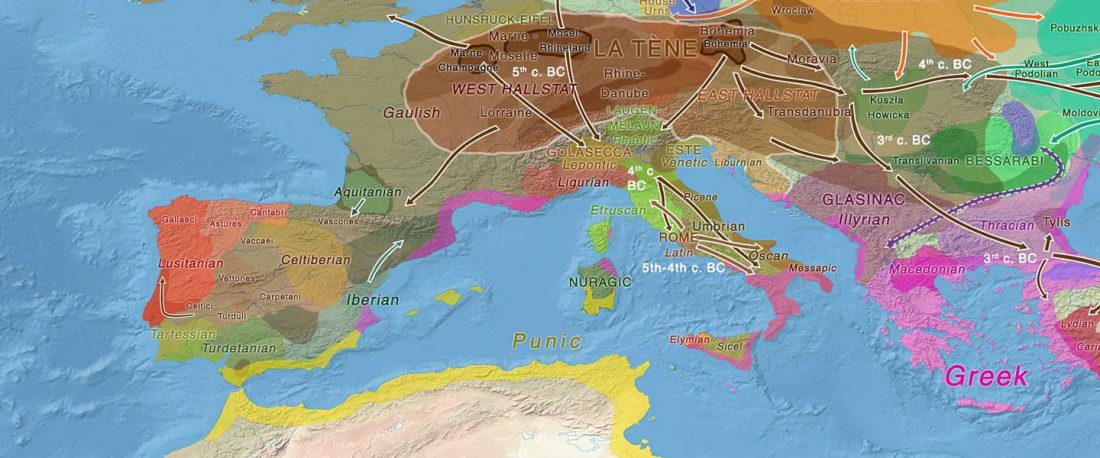
- Iron Age to Medieval Y-DNA
- Iron Age to Medieval Ancestry
- Iron Age to Medieval Y-DNA + Ancestry
- FTDNA’s big public debut