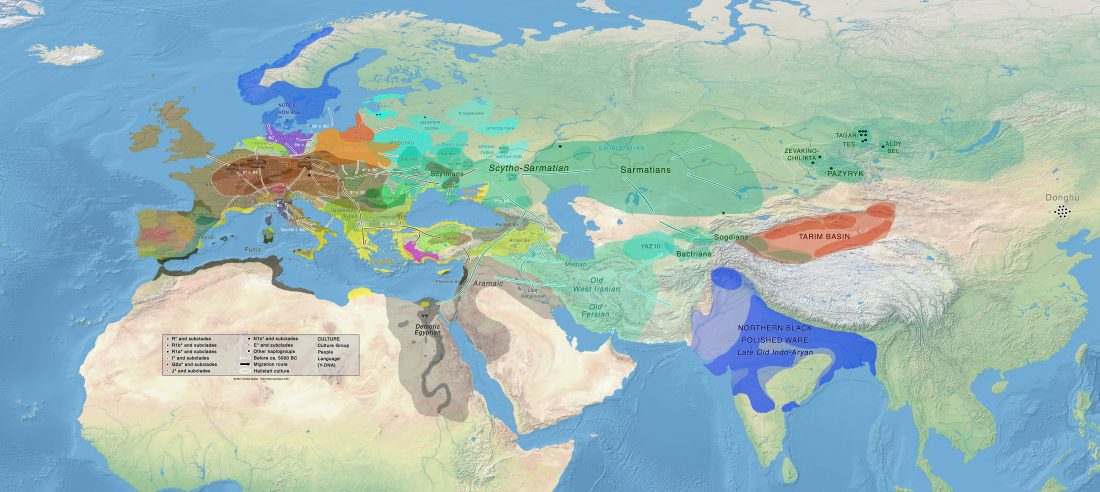
Two important papers have appeared regarding the supposed link of Uralians with haplogroup N.
Avars of haplogroup N1c-Tat
Preprint Genetic insights into the social organisation of the Avar period elite in the 7th century AD Carpathian Basin, by Csáky et al. bioRxiv (2019).
Interesting excerpts (emphasis mine):
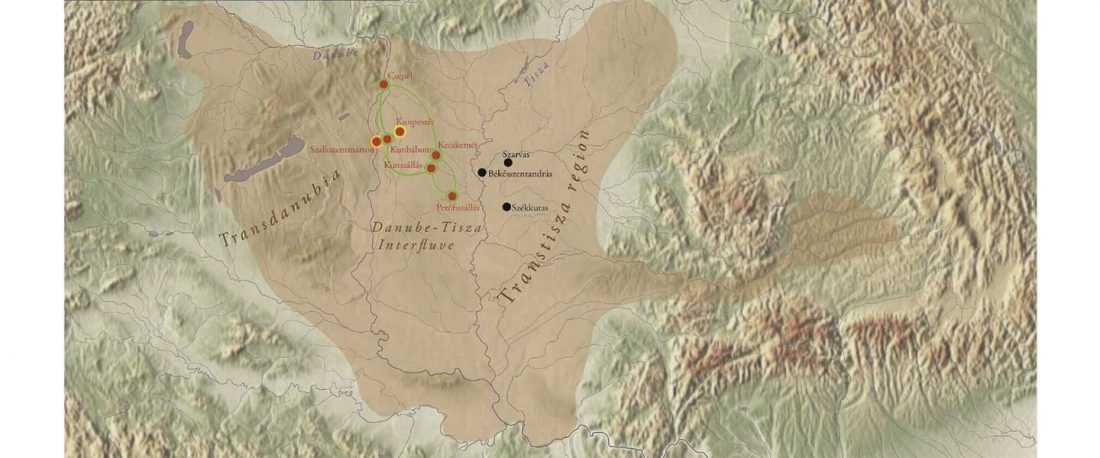
… Read the rest “R1a-Z280 and R1a-Z93 shared by ancient Finno-Ugric populations; N1c-Tat expanded with Micro-Altaic”After 568 AD the Avars settled in the Carpathian Basin and founded the Avar Qaganate that was an important power in Central Europe until the 9th century. Part of the Avar society was probably of Asian origin, however the localisation of their homeland is hampered by the scarcity of historical and archaeological