Deep population history of North, Central and South America
Open access Reconstructing the Deep Population History of Central and South America, by Posth et al. Cell (2018).
Abstract:
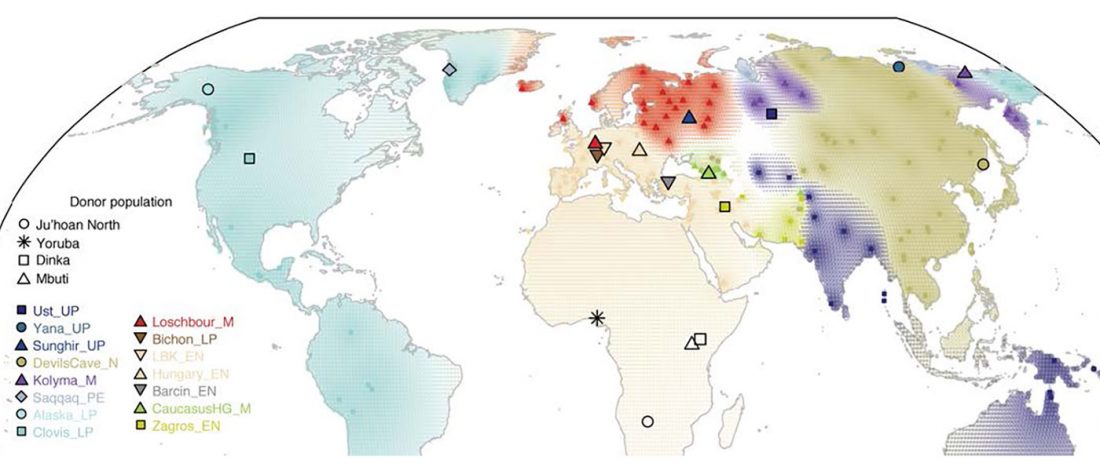
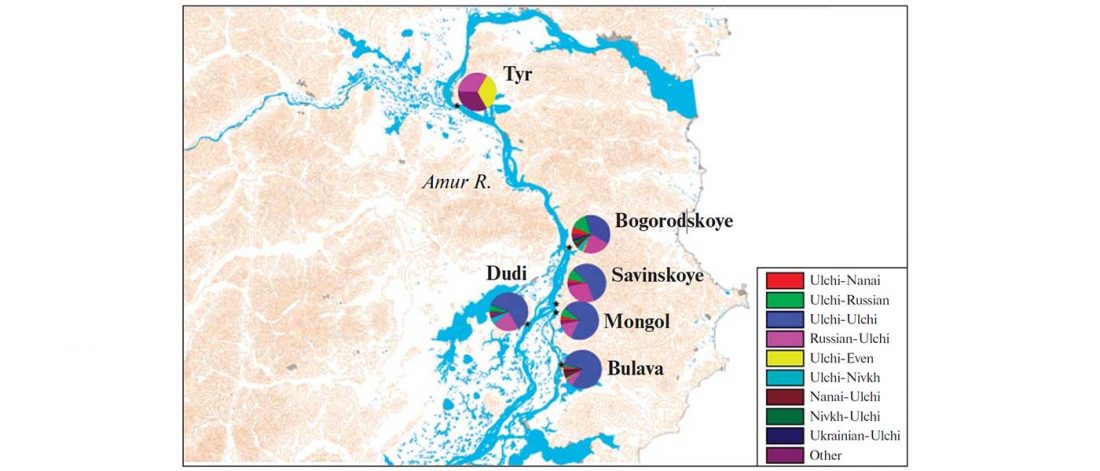
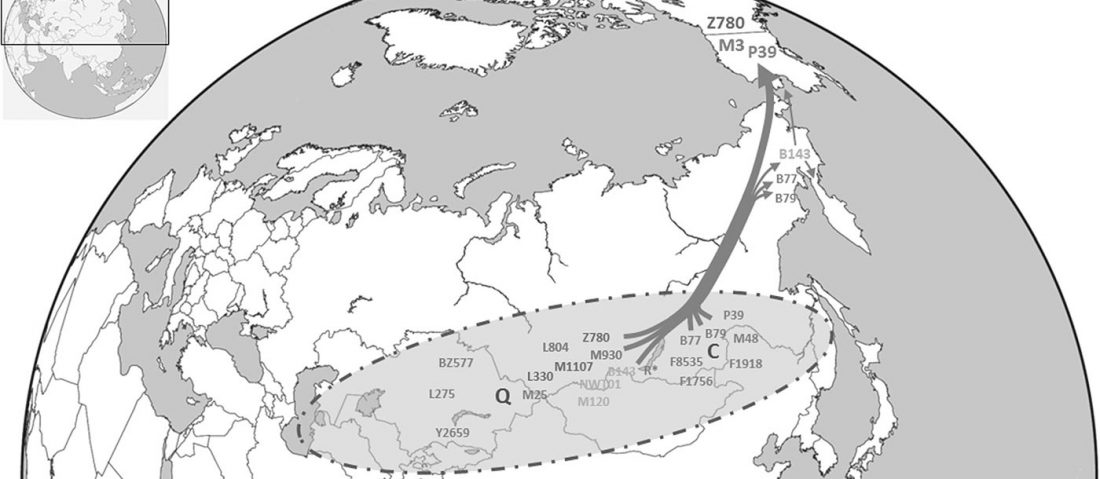
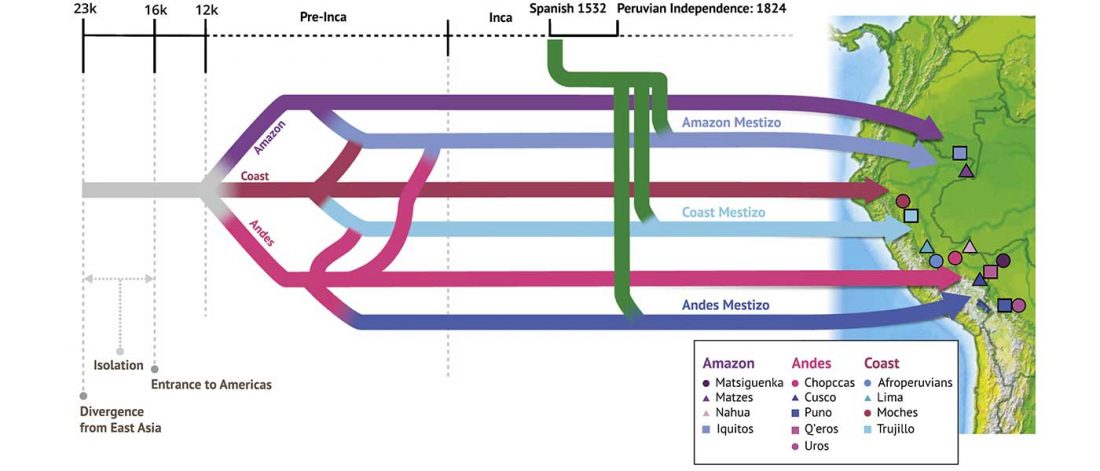
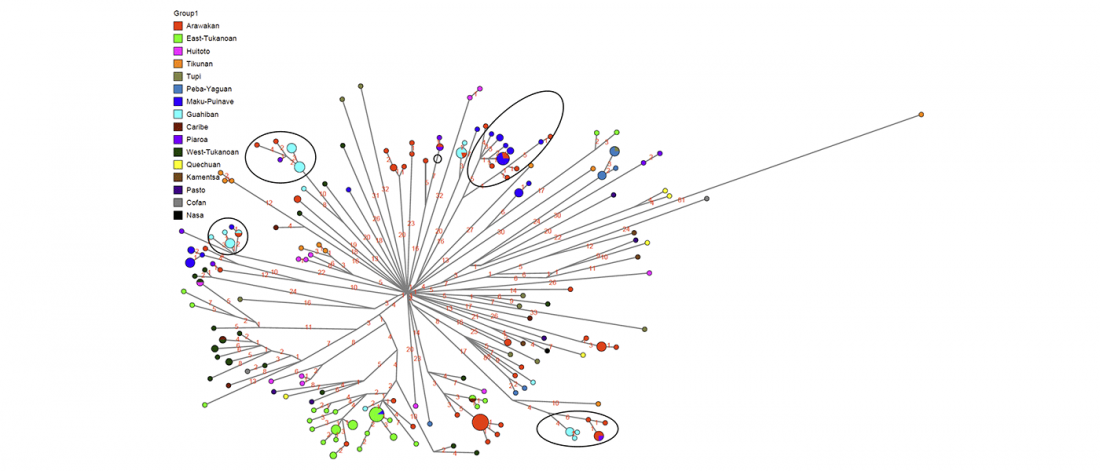
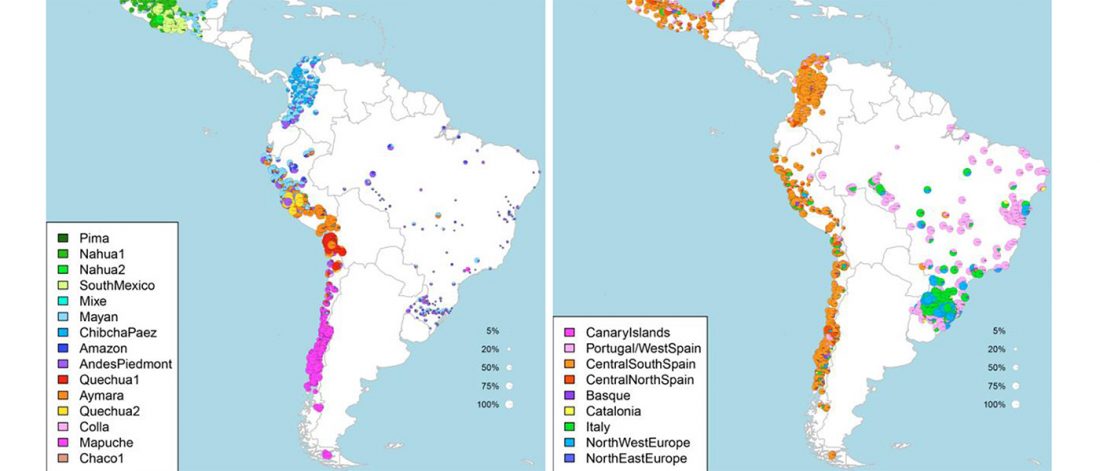
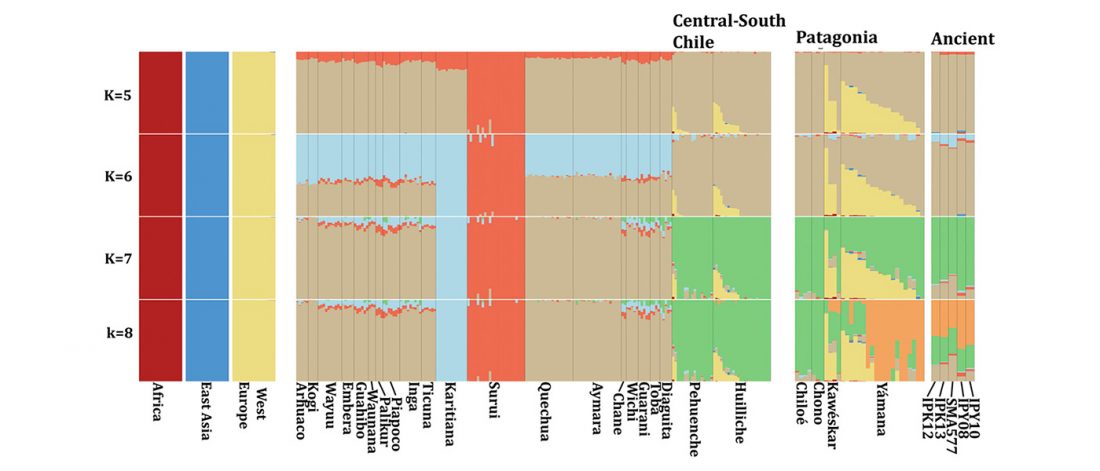
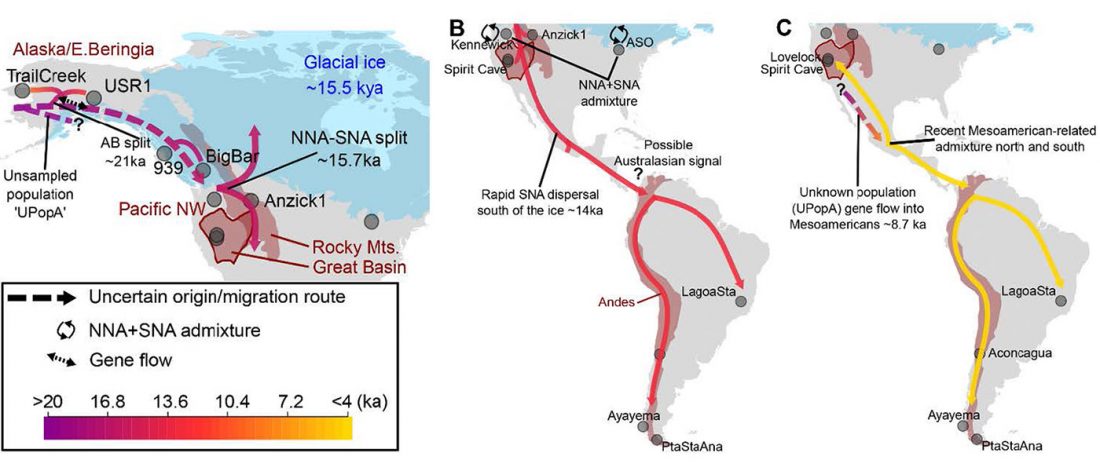
… Read the rest “Deep population history of North, Central and South America”We report genome-wide ancient DNA from 49 individuals forming four parallel time transects in Belize, Brazil, the Central Andes, and the Southern Cone, each dating to at least ∼9,000 years ago. The common ancestral population radiated rapidly from just one of the two early branches that contributed to Native Americans today. We document two previously unappreciated streams of gene flow between North and South America. One affected the Central Andes by ∼4,200 years ago, while the other explains an affinity