Open access Detecting the Population Structure and Scanning for Signatures of Selection in Horses (Equus caballus) From Whole-Genome Sequencing Data, by Zhang et al, Evolutionary Bioinformatics (2018) 14:1–9.
Abstract (emphasis mine):
Animal domestication gives rise to gradual changes at the genomic level through selection in populations. Selective sweeps have been traced in the genomes of many animal species, including humans, cattle, and dogs. However, little is known regarding positional candidate genes and genomic regions that exhibit signatures of selection in domestic horses. In addition, an understanding of the genetic processes underlying horse domestication, especially the origin of Chinese native populations, is still lacking. In our study, we generated whole genome sequences from 4 Chinese native horses and combined them with 48 publicly available full genome sequences, from which 15 341 213 high-quality unique single-nucleotide polymorphism variants were identified. Kazakh and Lichuan horses are 2 typical Asian native breeds that were formed in Kazakh or Northwest China and South China, respectively. We detected 1390 loss-of-function (LoF) variants in protein-coding genes, and gene ontology (GO) enrichment analysis revealed that some LoF-affected genes were overrepresented in GO terms related to the immune response. Bayesian clustering, distance analysis, and principal component analysis demonstrated that the population structure of these breeds largely reflected weak geographic patterns. Kazakh and Lichuan horses were assigned to the same lineage with other Asian native breeds, in agreement with previous studies on the genetic origin of Chinese domestic horses. We applied the composite likelihood ratio method to scan for genomic regions showing signals of recent selection in the horse genome. A total of 1052 genomic windows of 10 kB, corresponding to 933 distinct core regions, significantly exceeded neutral simulations. The GO enrichment analysis revealed that the genes under selective sweeps were overrepresented with GO terms, including “negative regulation of canonical Wnt signaling pathway,” “muscle contraction,” and “axon guidance.” Frequent exercise training in domestic horses may have resulted in changes in the expression of genes related to metabolism, muscle structure, and the nervous system.
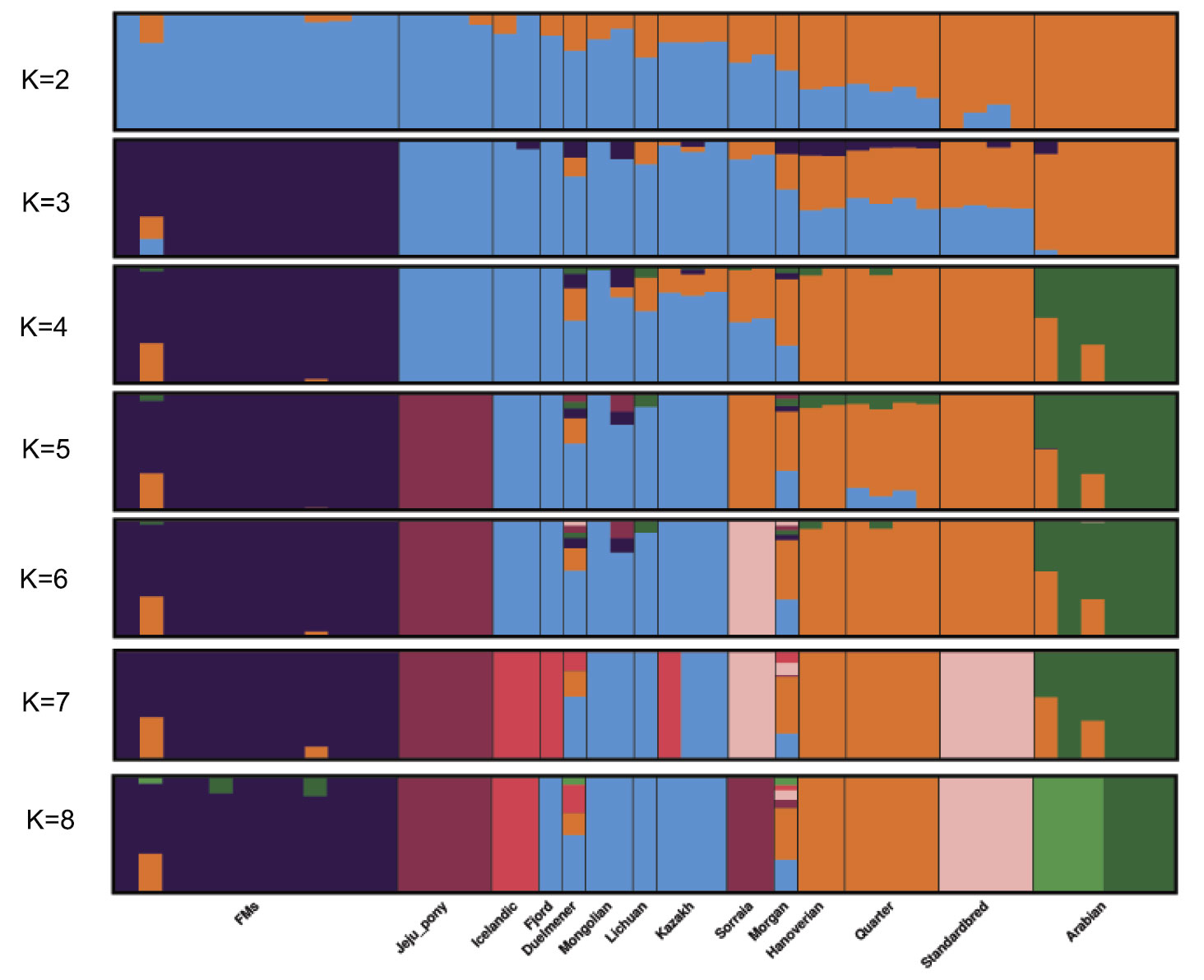
Interesting excerpts:
Admixture proportions were assessed without user-defined population information to infer the presence of distinct populations among the samples (Figure 2). At K = 3 or K = 4, Franches-Montagnes and Arabian forms one unique cluster; at K = 5, Jeju pony forms one unique cluster. For other breeds, comparatively strong population structure exists among breeds, and they can be assigned to 2 (or 3) alternate clusters from K = 3 to K = 5 including group A (Duelmener, Fjord, Icelandic, Kazakh, Lichuan, and Mongolian) and group B (Hanoverian, Morgan, Quarter, Sorraia, and Standardbred). For group A, geographically this was unexpected, where Nordic breeds (Norwegian Fjord, Icelandic, and Duelmener) clustered with Asian breeds including the Mongolian. Previous results of mitochondrial DNA have revealed links between the Mongolian horse and breeds in Iceland, Scandinavia, Central Europe, and the British Isles. The Mongol horses are believed to have been originally imported from Russia subsequently became the basis for the Norwegian Fjord horse.31 At K = 6, Sorraia forms one unique cluster. The Sorraia horse has no long history as a domestic breed but is considered to be of a nearly ancestral type in the southern part of the Iberian Peninsula.32 However, our result did not support Sorraia as an independent ancestral type based on result from K = 2 to K = 5, and the unique cluster in K = 6 may be explained by the small population size and recently inbreeding programs. Genetic admixture of Morgan reveals that these breeds are currently or traditionally continually crossed with other breeds from K = 2 to K = 8. The Morgan horse has been a largely closed breed for 200 years or more but there has been some unreported crossbreeding in recent times.33
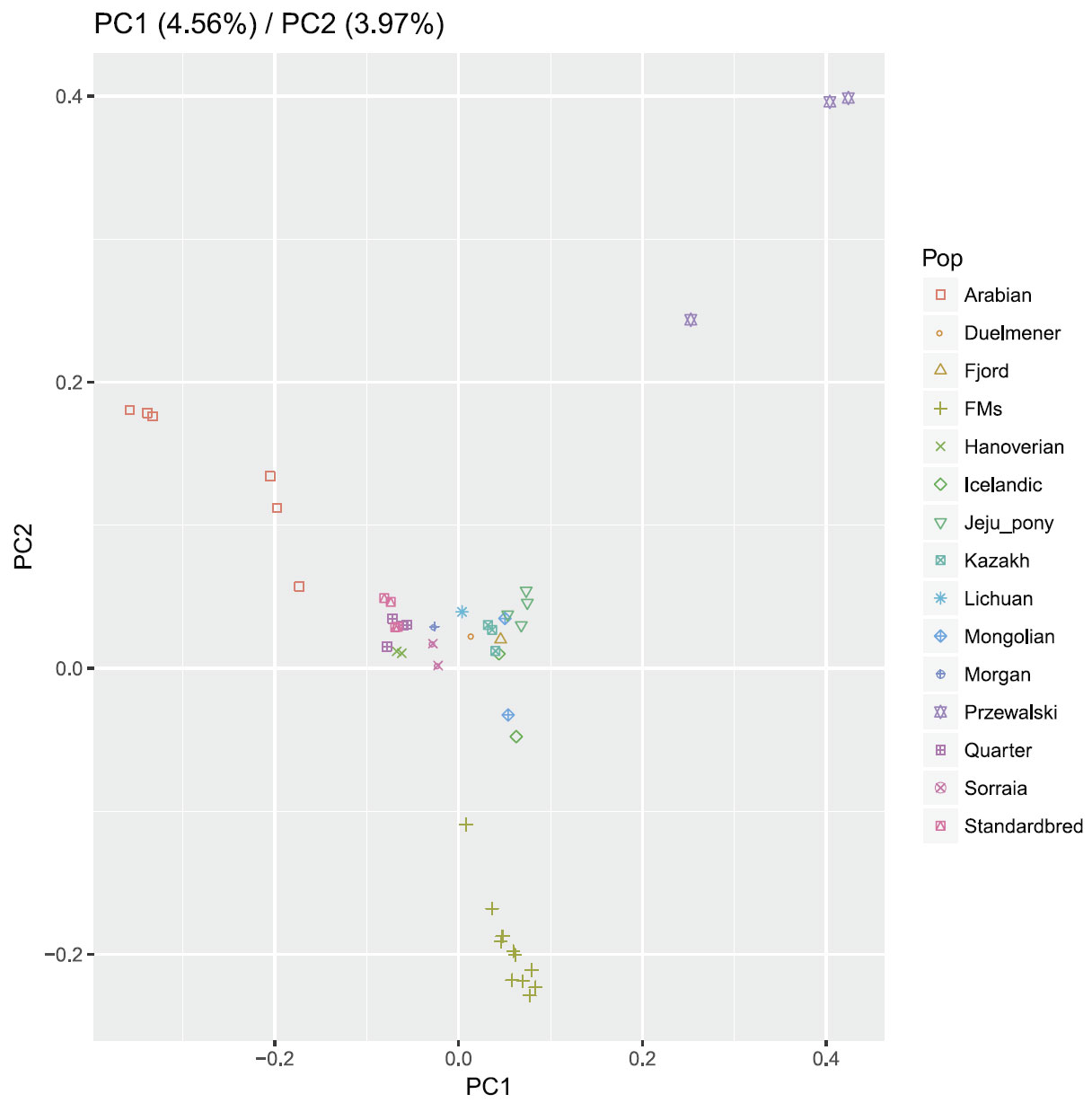
Bayesian clustering and PCA demonstrated the relationships among the horse breeds with weak geographic patterns. The tight grouping within most native breeds and looser grouping of individuals in admixed breeds have been reported previously in modern horses using data from a 54K SNP chip.33,34 Cluster analysis reveals that Arabian or Franches-Montagnes forms one unique cluster with relatively low K value, which is consistent with former study using 50K SNP chip 33,34 Interestingly, Standardbred forms a unique cluster with relatively high K value in this study, different from previous study.33 To date, no footprints are available to describe how the earliest domestic horses spread into China in ancient times. Our study found that Kazakh and Lichuan were assigned to the same lineage as other native Asian breeds, in agreement with previous studies on the origin of Chinese domestic horses.4,5,35,36 The strong genetic relationship between Asian native breeds and European native breeds have made it more difficult to understand the population history of the horse across Eurasia. Low levels of population differentiation observed between breeds might be explained by historical admixture. Unlike the domestic pig in China,8 we suggest that in China, Northern/Southern distinct groups could not be used to genetically distinct native Chinese horse breeds. We consider that during domestication process of horse, gene flow continued among Chinese-domesticated horses.
Open access Some maternal lineages of domestic horses may have origins in East Asia revealed with further evidence of mitochondrial genomes and HVR-1 sequences, by Ma et al., PeerJ (2018).
Abstract:
Objectives
There are large populations of indigenous horse (Equus caballus) in China and some other parts of East Asia. However, their matrilineal genetic diversity and origin remained poorly understood. Using a combination of mitochondrial DNA (mtDNA) and hypervariable region (HVR-1) sequences, we aim to investigate the origin of matrilineal inheritance in these domestic horses.Methods
To investigate patterns of matrilineal inheritance in domestic horses, we conducted a phylogenetic study using 31 de novo mtDNA genomes together with 317 others from the GenBank. In terms of the updated phylogeny, a total of 5,180 horse mitochondrial HVR-1 sequences were analyzed.Results
Eighteen haplogroups (Aw-Rw) were uncovered from the analysis of the whole mitochondrial genomes. Most of which have a divergence time before the earliest domestication of wild horses (about 5,800 years ago) and during the Upper Paleolithic (35–10 KYA). The distribution of some haplogroups shows geographic patterns. The Lw haplogroup contained a significantly higher proportion of European horses than the horses from other regions, while haplogroups Jw, Rw, and some maternal lineages of Cw, have a higher frequency in the horses from East Asia. The 5,180 sequences of horse mitochondrial HVR-1 form nine major haplogroups (A-I). We revealed a corresponding relationship between the haplotypes of HVR-1 and those of whole mitochondrial DNA sequences. The data of the HVR-1 sequences also suggests that Jw, Rw, and some haplotypes of Cw may have originated in East Asia while Lw probably formed in Europe.Conclusions
Our study supports the hypothesis of the multiple origins of the maternal lineage of domestic horses and some maternal lineages of domestic horses may have originated from East Asia.
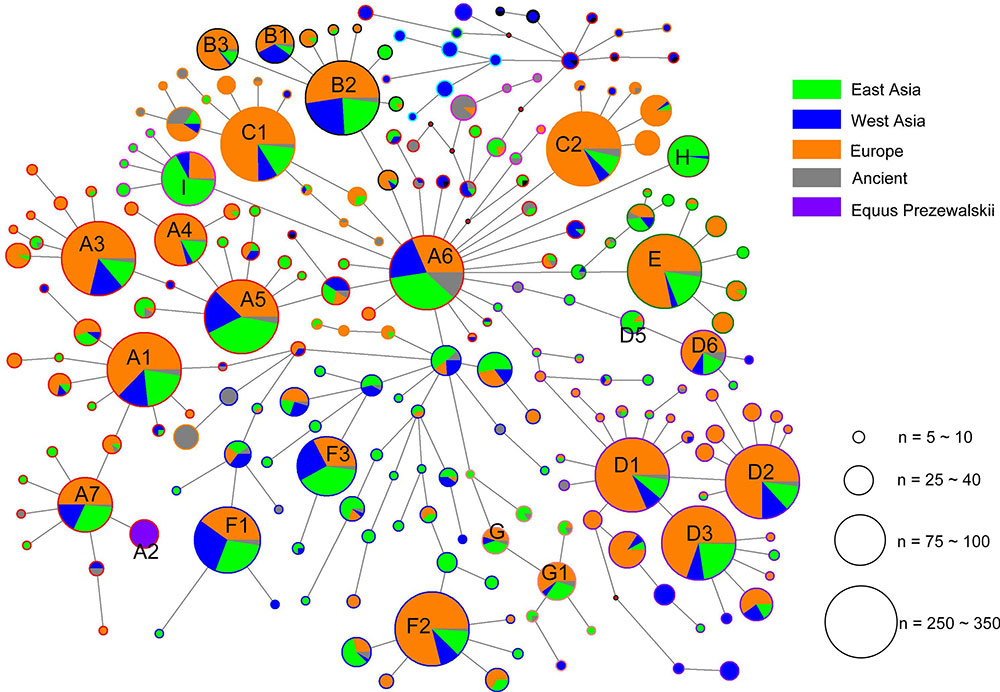
Geographic distributions of horse mtDNA haplogroups
The analysis of geographic distribution of the mitochondrial genome haplogroups showed that horse populations in Europe or East Asia included all haplogroups defined from the mtDNA genome sequences. The lineage Fw comprised entirely of Przewalskii horses. The two haplogroups Iw and Lw displayed frequency peaks in Europe (14.08% and 37.32%, respectively) and a decline to the east (9.33% and 8.00% in the West Asia, and 6.45% and 12.90% in East Asia, respectively), especially for Lw, which contained the largest number of European horses (Table 2). However, an opposite distribution pattern was observed for haplogroups Aw, Hw, Jw, and Rw, which were harbored by more horses from East Asia than those from other regions. The proportions of horses from East Asia for the four haplogroups were 38%, 88%, 62%, and 54%, respectively.
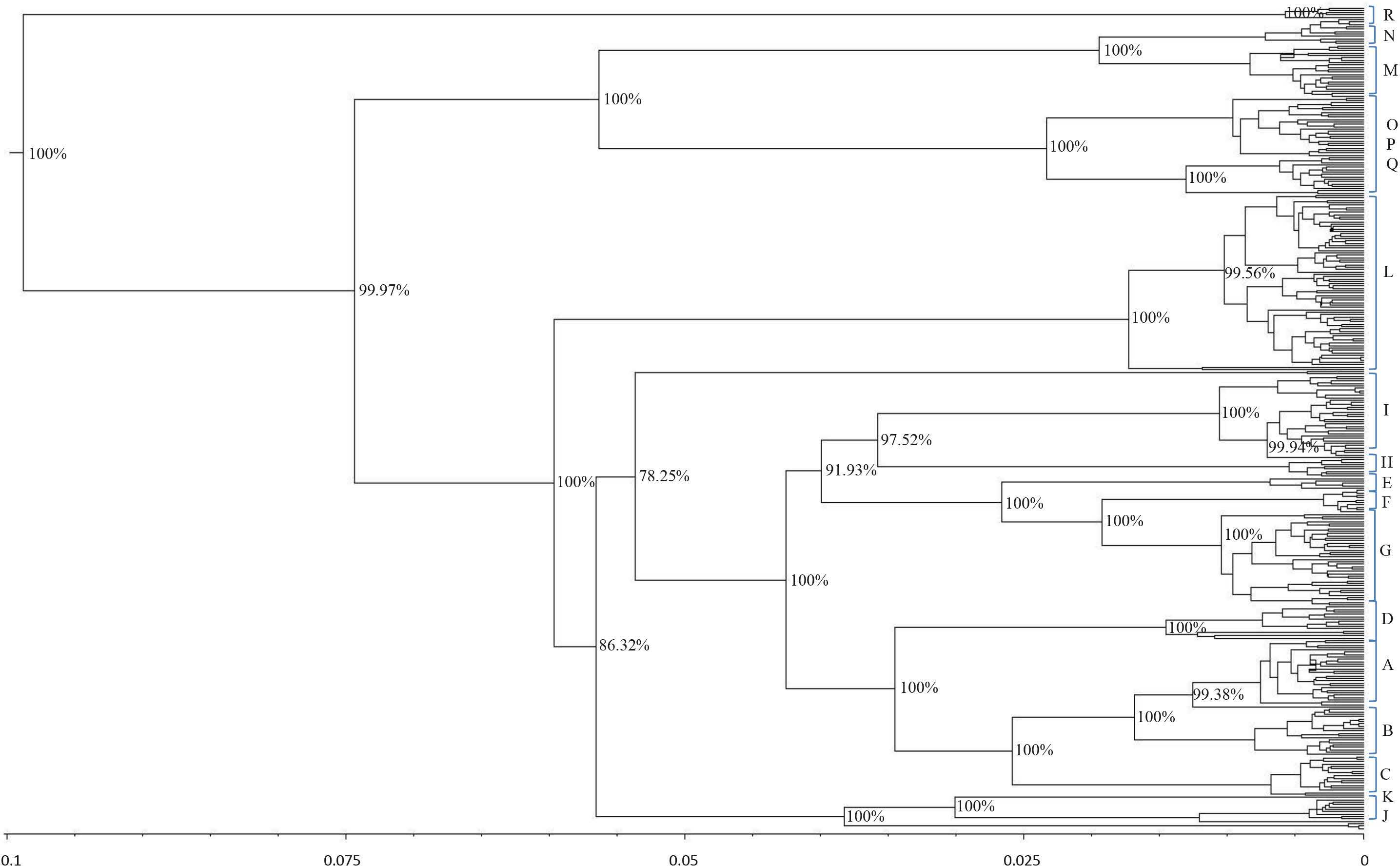
and was rooted at a donkey (E. asinus) mitochondrial genome (not displayed). The topology was inferred by a beast approach, whereas a time divergence scale (based on rate substitutions) is shown on the bottom (age estimates were indicated with thousand years (KY)). The percentages on each branch represent Bayesian posterior credibility and the alphabets on the right represent the names of haplogroups. Additional details concerning ages were given in Tables S3 and S6.
Related:
- Ancient DNA upends the horse family tree
- Consequences of Damgaard et al. 2018 (I): EHG ancestry in Maykop samples, and the potential Anatolian expansion routes
- No large-scale steppe migration into Anatolia; early Yamna migrations and MLBA brought LPIE dialects in Asia
- Domestication spread probably via the North Pontic steppe to Khvalynsk… but not horse riding
- Differing modes of animal exploitation in North Pontic Eneolithic and Bronze Age Societies
- North Pontic steppe Eneolithic cultures, and an alternative Indo-Slavonic model
- The Lower Danube during the Eneolithic, and the potential Proto-Anatolian community
- Eneolithic Ukraine cultures of the North Pontic steppe and southern steppe-forest, on the Left Bank of the Dnieper
- Recent archaeological finds near Indo-European and Uralic homelands
- Proto-Indo-European homeland south of the Caucasus?
- On the potential origin of Caucasus hunter-gatherer ancestry in Eneolithic steppe cultures
- Consequences of O&M 2018 (II): The unsolved nature of Suvorovo-Novodanilovka chiefs, and the route of Proto-Anatolian expansion
- New Ukraine Eneolithic sample from late Sredni Stog, near homeland of the Corded Ware culture