Open access study Cultural hitchhiking and competition between patrilineal kin groups explain the post-Neolithic Y-chromosome bottleneck, by Zeng, Aw, and Feldman, Nature Communications (2018).
Abstract (emphasis mine):
In human populations, changes in genetic variation are driven not only by genetic processes, but can also arise from cultural or social changes. An abrupt population bottleneck specific to human males has been inferred across several Old World (Africa, Europe, Asia) populations 5000–7000 BP. Here, bringing together anthropological theory, recent population genomic studies and mathematical models, we propose a sociocultural hypothesis, involving the formation of patrilineal kin groups and intergroup competition among these groups. Our analysis shows that this sociocultural hypothesis can explain the inference of a population bottleneck. We also show that our hypothesis is consistent with current findings from the archaeogenetics of Old World Eurasia, and is important for conceptions of cultural and social evolution in prehistory.
Relevant excerpts:
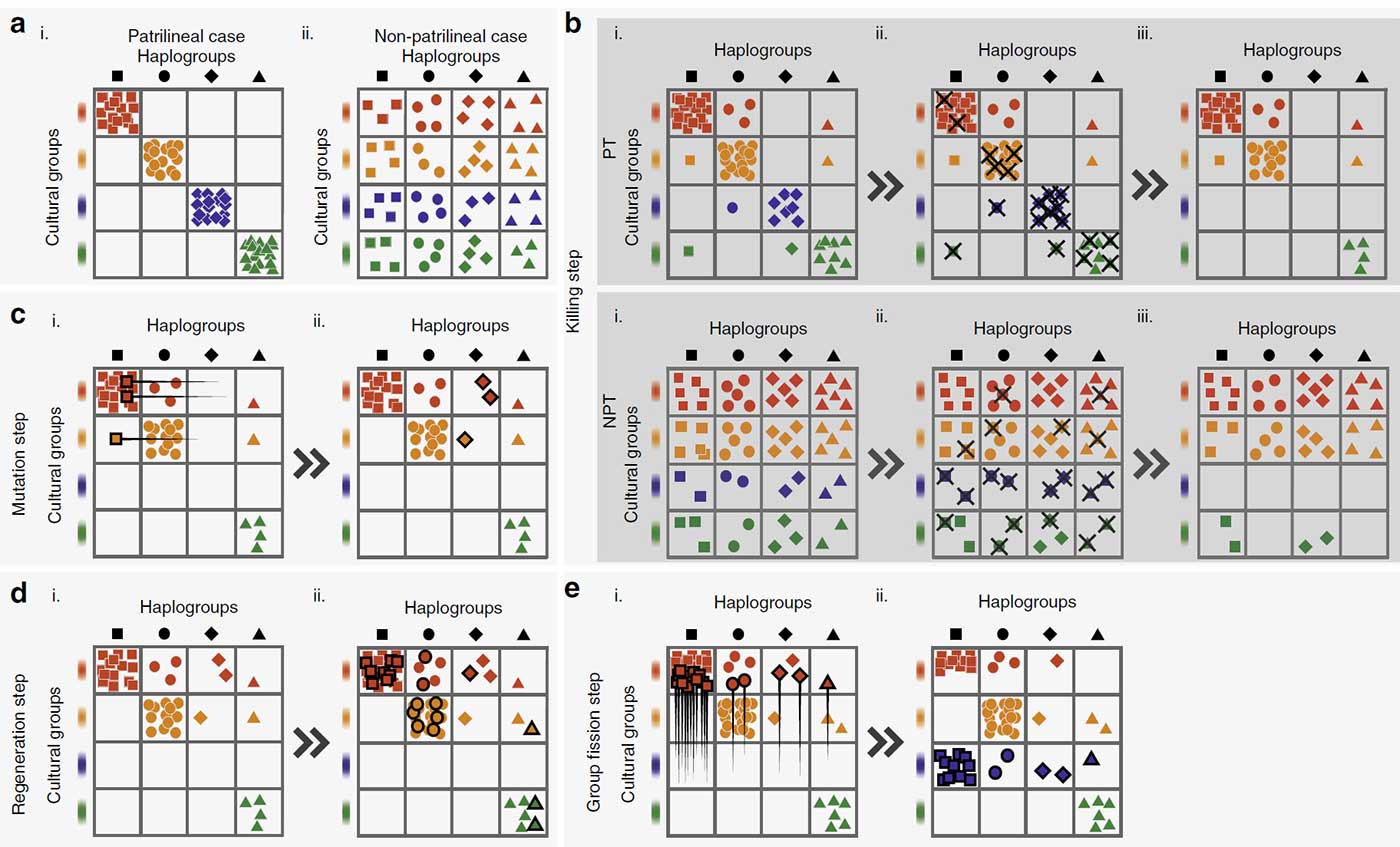
Our hypothesis explains the bottleneck as a consequence of intergroup competition between patrilineal kin groups, which caused cultural hitchhiking between Y-chromosomes and cultural groups and reduction in Y-chromosomal diversity. Competition between demes can dramatically reduce genetic diversity within a population1, especially if the population is structured such that variation is greater between demes than within demes. Culturally transmitted kinship ideals and norms can cause homophilous sorting and limit interdemic gene flow, creating homogeneous demes that differ strongly from one another. Patrilineal corporate kin groups, with coresiding male group members descending from a common male ancestor, would produce such an effect on Y-chromosomes only, as patrilineal corporate kin groups generally coexist with female exogamy40, which would homogenize the mitochondrial gene pools of different groups41,42.
With intergroup competition between patrilineal corporate kin groups, two mechanisms would operate to reduce Y-chromosomal diversity. First, patrilineal corporate kin groups produce high levels of Y-chromosomal homogeneity within each social group due to common descent, as well as high levels of between-group variation. Second, the presence of such groups results in violent intergroup competition preferentially taking place between members of male descent groups, instead of between unrelated individuals. Casualties from intergroup competition then tend to cluster among related males, and group extinction is effectively the extinction of lineages.
There is evidence that other analogous situations involving gene-culture hitchhiking in culturally-defined social groups may have affected genetic diversity. Central Asian pastoralists, who are organized into patriclans, have high levels of intergroup competition and demonstrate ethnolinguistic and population-genetic turnover down into the historical period59. They also have a markedly lower diversity in Y-chromosomal lineages than nearby agriculturalists42,60. In fact, Central Asians are the only population whose male effective population size has not recovered from the post-Neolithic bottleneck; it remains disproportionately reduced, compared to female estimates using mtDNA4. Central Asians are also the only population to have star-shaped expansions of Y-chromosomes within the historical period, which may be due to competitive processes that led to the disproportionate political success of certain patrilineal clans60.
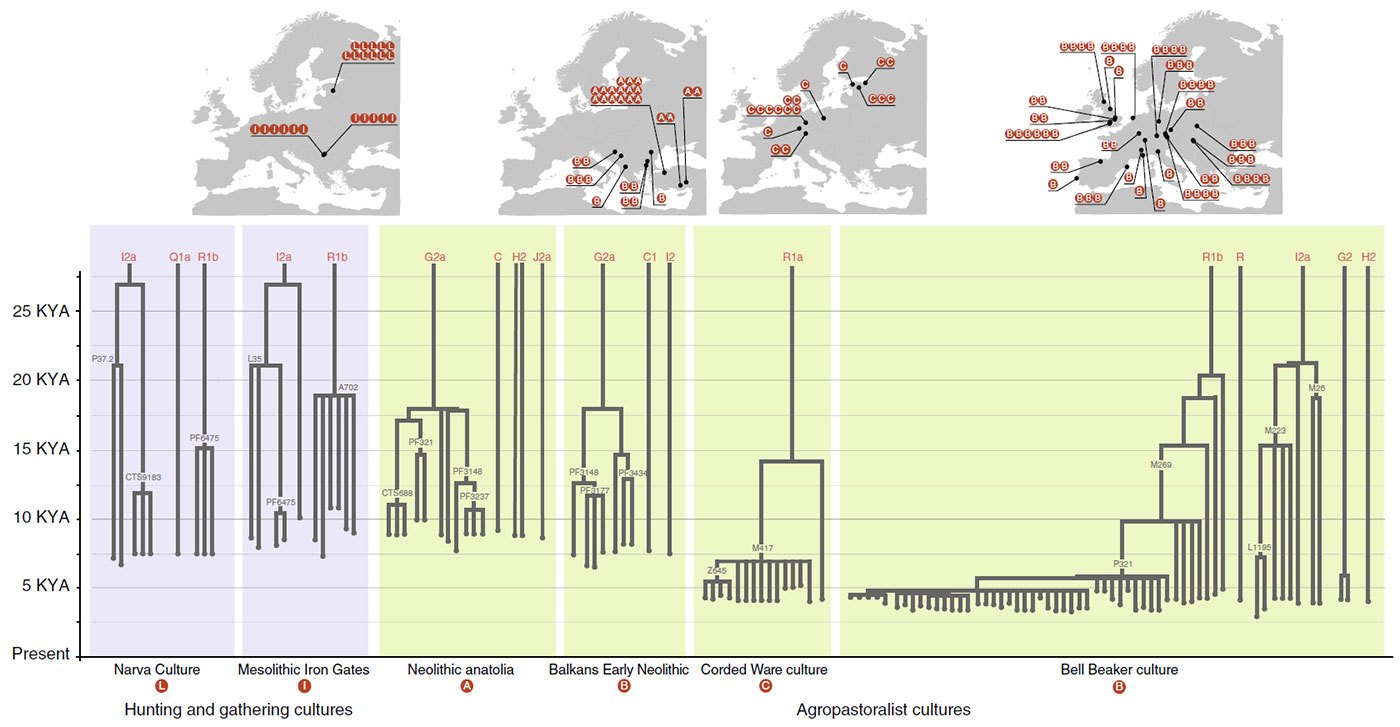
The simulation offers an interesting graphic. I had been thinking for some time about developing an interactive image with waves of expansion showing how only few haplogroups expand and thus their variability is reduced in successive migration waves, because a lot of people seemed not to be willing to accept this:
You only have to imagine this process happening in many successive waves of expansion (external as well as internal to each culture) since the first Neolithic expansions in the steppe in the late-6th millennium BC, even before the formation of the Khvalynsk-Sredni Stog cultural-historical community, to understand what happened in the next thousands of years with evolving patrilineal clans and their distinct cultures.
The whole paper is an interesting read. It’s great to see sociology and genetics finally catch up and interact to develop more complex anthropological hypotheses.
The fact that this paper appears in mid-2018 and geneticists are beginning to discuss this only now when their statistical methods fail to explain the obvious (see David Reich’s recent interview) seems anachronistic, though, because all this was quite clear already in 2015 – at least for those who were looking for mainstream Yamna – Bell Beaker connections, instead of inventing new migration pathways to justify the results of certain statistical analyses…
Anyway, better late than never.
Also, they use YFull estimates, which vindicates my use of them in the Indo-European demic diffusion model (2017). On the other hand, their use of these estimates right now in 2018 for R1a-M417 and R1b-M269 – when we know of a R1a-Z93 case much older than YFull’s estimated 5,000 YBP for this subclade, and possibly for R1b-L23, too, is the biggest pitfall in their temporal assessment, although the bottlenecks seen in Chalcolithic expansions seem to have indeed began during the Mesolithic-Neolithic transition in the steppe.
So, say goodbye (if you haven’t already) to dat fantasy ‘steppe people’ of mixed R1a/R1b descent cooperating with the same mixed steppe language, all represented by the Yamnaya™ ancestral component, and say hello to distinct, competing ethnolinguistic steppe groups during the Neolithic.
Related:
- Haplogroup is not language, but R1b-L23 expansion was associated with Proto-Indo-Europeans
- Lazaridis’ evolutionary history of human populations in Europe
- David Reich on social inequality and Yamna expansion with few Y-DNA subclades
- Olalde et al. and Mathieson et al. (Nature 2018): R1b-L23 dominates Bell Beaker and Yamna, R1a-M417 resurges in East-Central Europe during the Bronze Age
- The Indo-European demic diffusion model, and the “R1b – Indo-European” association