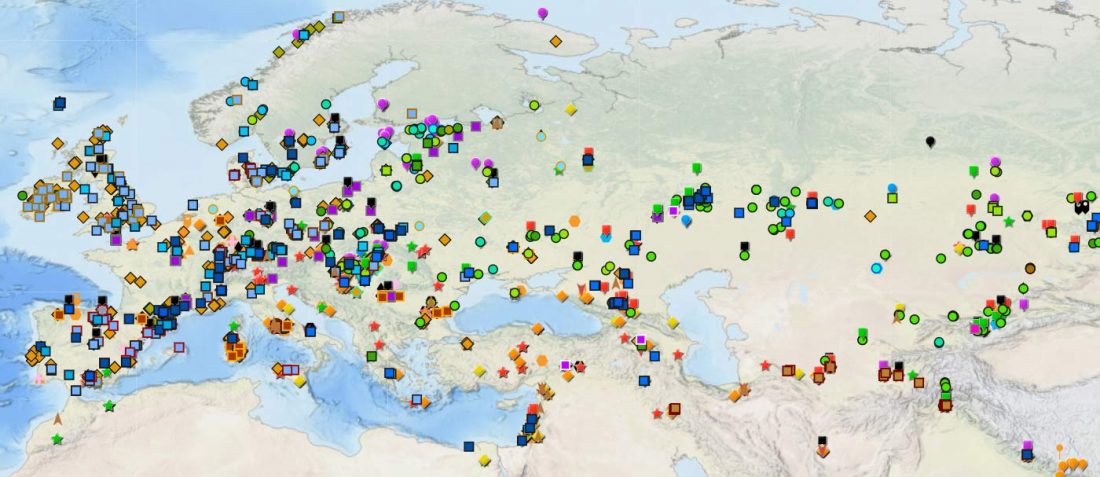
The Reich Lab has recently pre-published high quality shotgun sequencing data from 216 ancient individuals within the framework of the Allen Ancient Genome Diversity Project / John Templeton Ancient DNA Atlas. Metadata for the 216 genomes are available here.
Their median coverage is 4.9x, and among them there are 50 high coverage genomes (17-36x), but there are also samples with a coverage similar to the previously published ones.
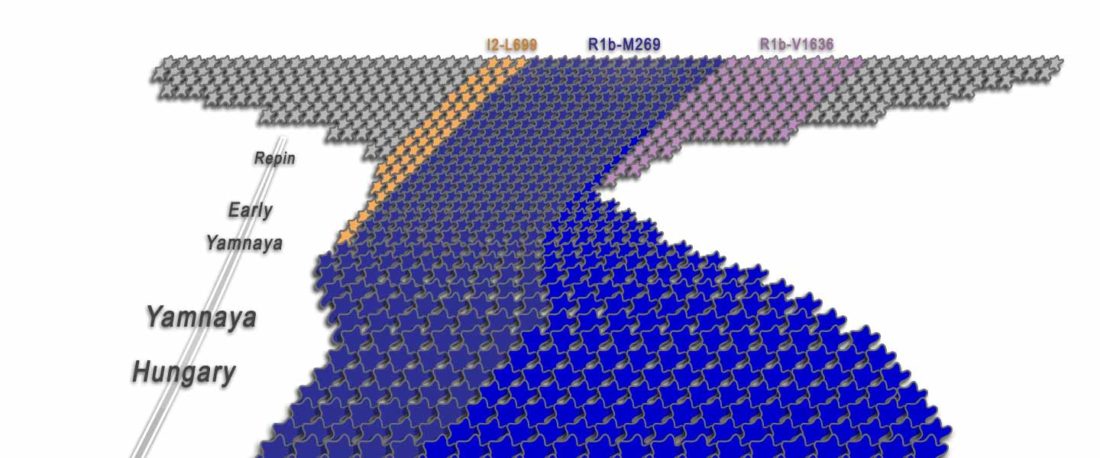
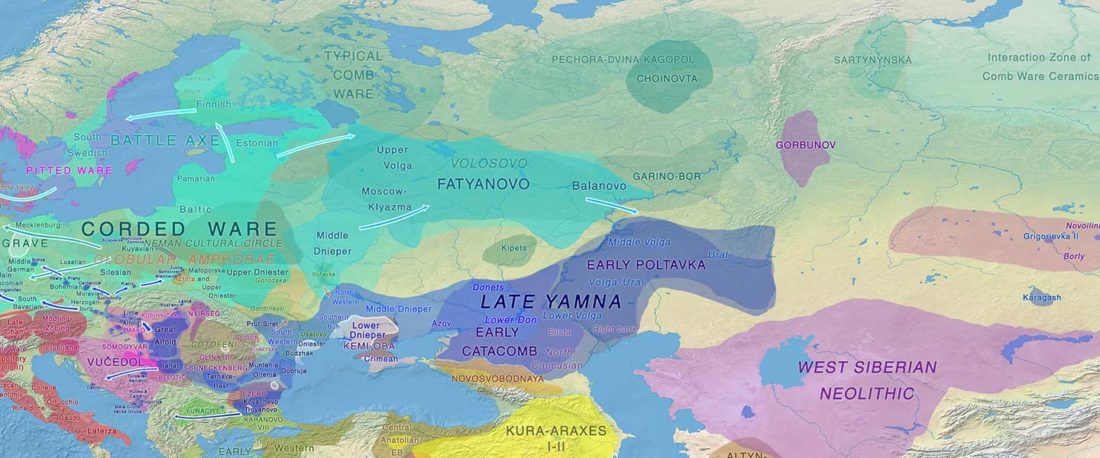
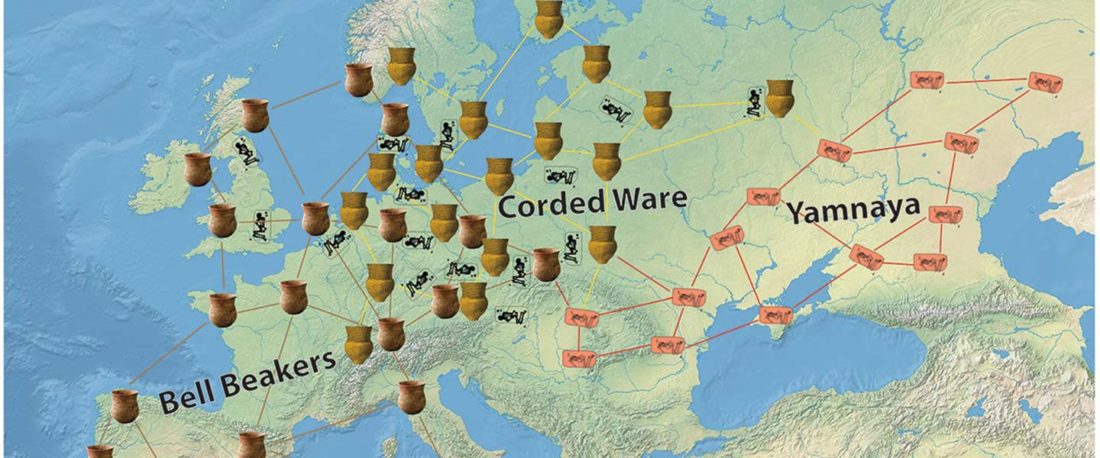
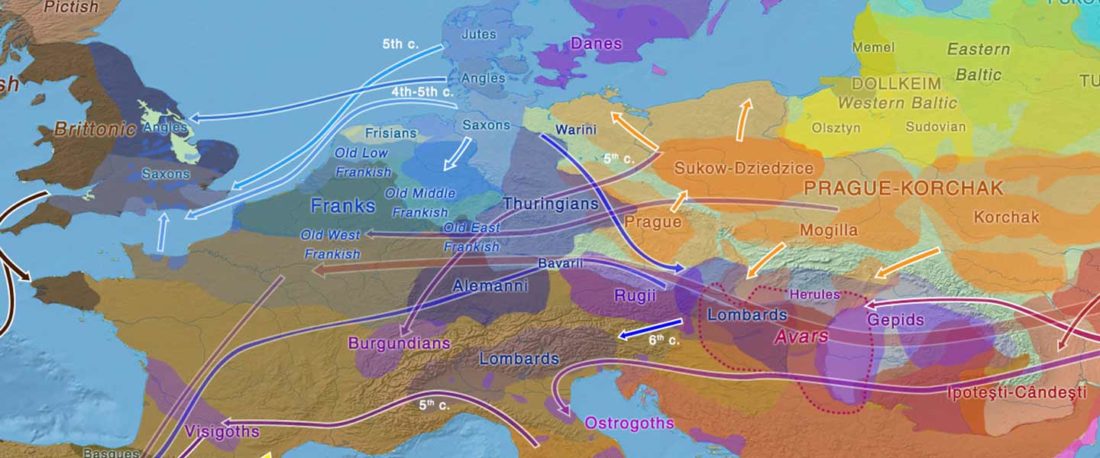
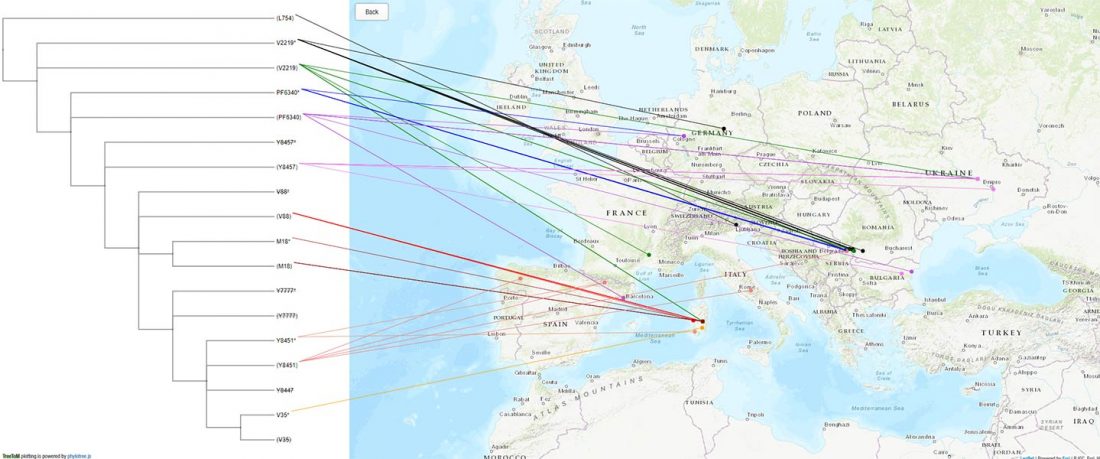
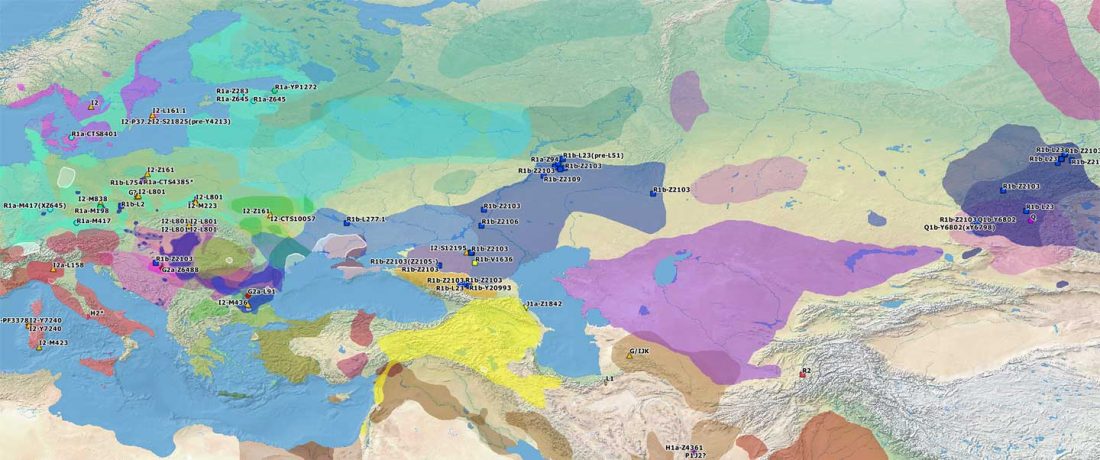
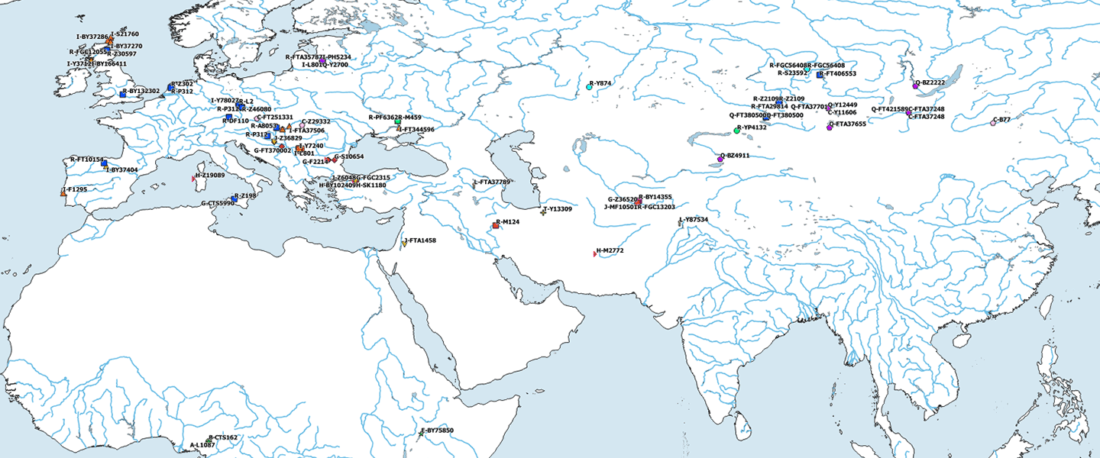
The FamilyTreeDNA Haplotree team formed by phylogeneticist Michael Sager and Göran Runfeldt from the R&D team has analyzed all 129 males for Y-SNP calls, using – and updating with them – … Read the rest “Y-DNA of 129 high quality shotgun ancient samples”