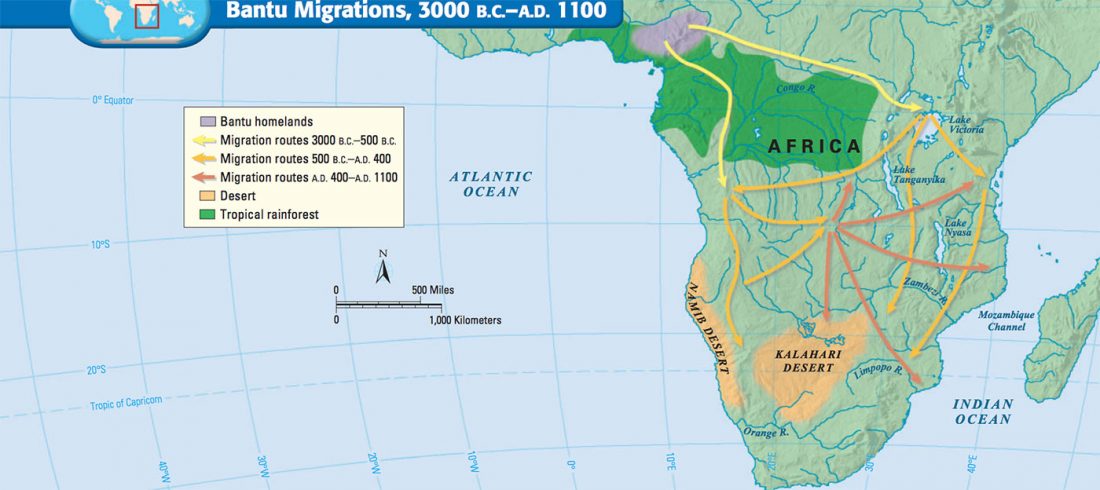
The role of matrilineality in shaping patterns of Y chromosome and mtDNA sequence variation in southwestern Angola, by Oliveira et al. bioRxiv (2018).
Interesting excerpts (emphasis mine):
The origins of NRY diversity in SW Angola
… Read the rest “Bantu distinguished from Khoe by uniparental markers, not genome-wide autosomal admixture”In accordance with our previous mtDNA study9, the present NRY analysis reveals a major division between the Kx’a-speaking !Xun and the Bantu-speaking groups, whose paternal genetic ancestry does not display any old remnant lineages, or a clear link to pre-Bantu eastern African migrants introducing Khoe-Kwadi languages and pastoralism into southern Africa (cf. 15). This is especially evident in the distribution of the