Open access Bayesian estimation of partial population continuity by using ancient DNA and spatially explicit simulations, by Silva et al., Evolutionary Applications (2018).
Abstract (emphasis mine):
The retrieval of ancient DNA from osteological material provides direct evidence of human genetic diversity in the past. Ancient DNA samples are often used to investigate whether there was population continuity in the settlement history of an area. Methods based on the serial coalescent algorithm have been developed to test whether the population continuity hypothesis can be statistically rejected by analysing DNA samples from the same region but of different ages. Rejection of this hypothesis is indicative of a large genetic shift, possibly due to immigration occurring between two sampling times. However, this approach is only able to reject a model of full continuity model (a total absence of genetic input from outside), but admixture between local and immigrant populations may lead to partial continuity. We have recently developed a method to test for population continuity that explicitly considers the spatial and temporal dynamics of populations. Here we extended this approach to estimate the proportion of genetic continuity between two populations, by using ancient genetic samples. We applied our original approach to the question of the Neolithic transition in Central Europe. Our results confirmed the rejection of full continuity, but our approach represents an important step forward by estimating the relative contribution of immigrant farmers and of local hunter‐gatherers to the final Central European Neolithic genetic pool. Furthermore, we show that a substantial proportion of genes brought by the farmers in this region were assimilated from other hunter‐gatherer populations along the way from Anatolia, which was not detectable by previous continuity tests. Our approach is also able to jointly estimate demographic parameters, as we show here by finding both low density and low migration rate for pre‐Neolithic hunter‐gatherers. It provides a useful tool for the analysis of the numerous aDNA datasets that are currently being produced for many different species.
Relevant excerpts:
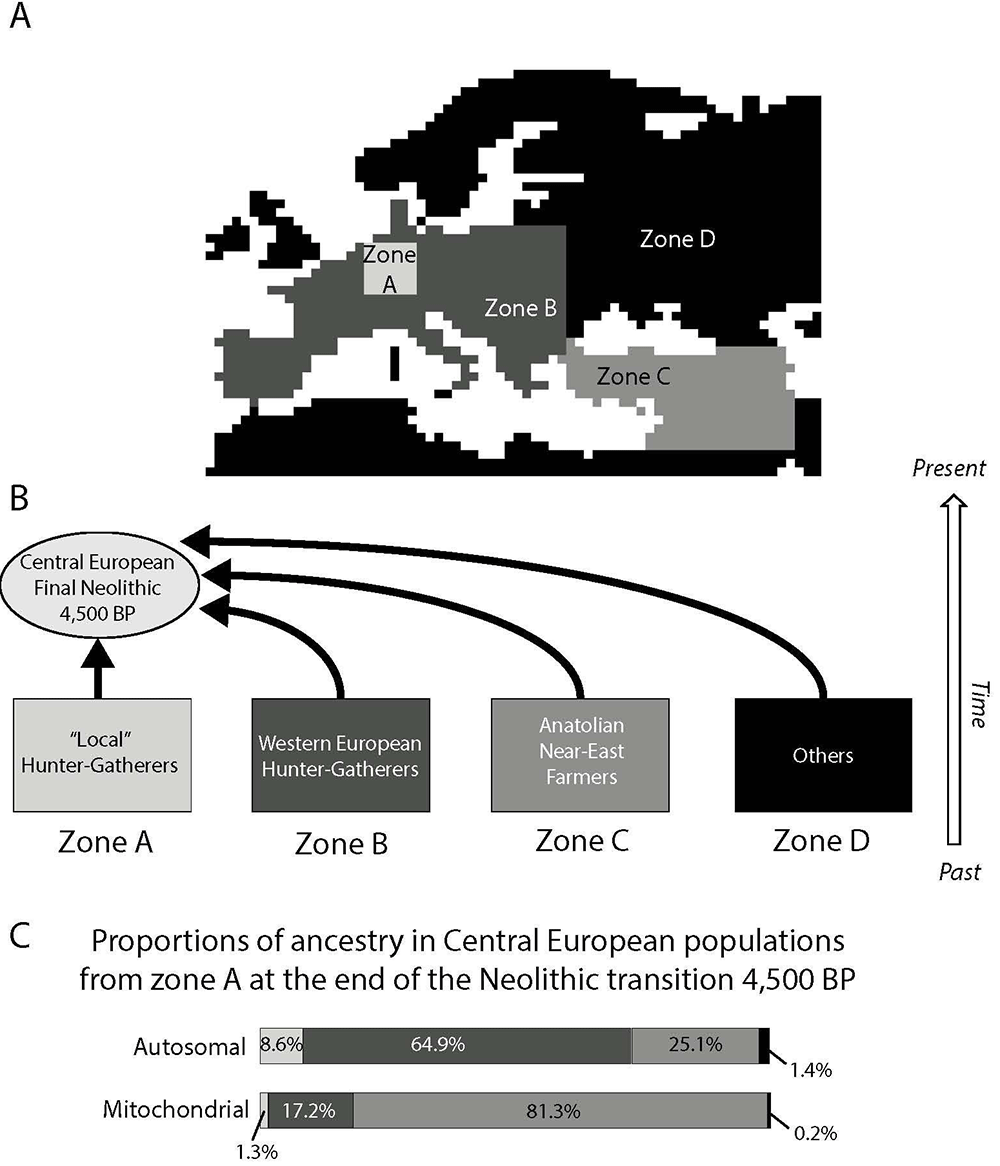
Our results are in general accordance with two distinct ancestry components that have previously been detected at the continental scale by Lazaridis, Patterson et al. (2014): the “early European farmer” (EEF), which corresponds here to the NFA from Anatolia (zone C in Figure 3), and the “West European hunter-gatherer” (WHG), which corresponds here to the PHG from zones A and B in Figure 3. Notably, the contribution of an Ancient North Eurasians (ANE) component is not included in our model as we did not consider potential post-Neolithic immigration waves, which could have contributed to the modern European genetic pool, such as the wave that came from the Pontic steppes and was associated with the Yamnaya culture (Haak, Lazaridis et al. 2015). Without considering the ANE ancestry component, our estimate of the autosomal genetic contribution of Early farmers to the gene pool of Central European populations (25%) tends to be lower than the EEF ancestry estimated in most modern Western European populations, but is of the same order than the estimations in modern Estonians and in the ancient Late Neolithic genome “Karsdorf” from Germany (Lazaridis, Patterson et al. 2014, Haak, Lazaridis et al. 2015). Note that the contribution of hunter-gatherers to Neolithic communities appears to be variable in different regions of Europe (Skoglund, Malmstrom et al. 2012, Brandt, Haak et al. 2013, Lazaridis, Patterson et al. 2014), while we computed an average value for Central Europe. Moreover, we computed the ancestry of the two groups at the end of the Neolithic period while previous studies estimated it in modern times. Finally, previous studies used molecular information to directly estimate admixture proportions, while we use molecular information to estimate the model parameters and, then, we computed the expected genetic contributions of both groups using the best parameters, without using molecular information during this second step. Model assumptions may thus influence the inferences on the relative genetic contribution of both groups. In particular, we made the assumption of a uniform expansion of NFA with constant and similar assimilation of PHG over the whole continent but spatio-temporally heterogeneous environment, variable assimilation rate and long distance dispersal may have played an important role. The effects of those factors should be investigated in future studies.