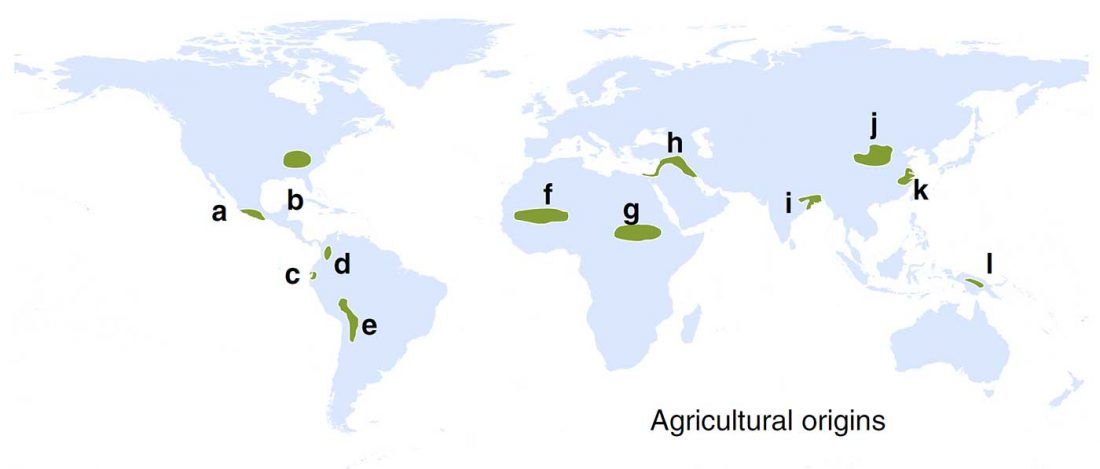
Improving environmental conditions favoured higher local population density, which favoured domestication
New paper (behind paywall) Hindcasting global population densities reveals forces enabling the origin of agriculture, by Kavanagh et al., Nature Human Behaviour (2018)
Abstract (emphasis mine):
… Read the rest “Improving environmental conditions favoured higher local population density, which favoured domestication”The development and spread of agriculture changed fundamental characteristics of human societies1,2,3. However, the degree to which environmental and social conditions enabled the origins of agriculture remains contested4,5,6. We test three hypothesized links between the environment, population density and the origins of plant and animal domestication, a prerequisite for agriculture: (1) domestication arose as environmental conditions improved and population densities increased7 (surplus hypothesis); (2) populations needed domestication to