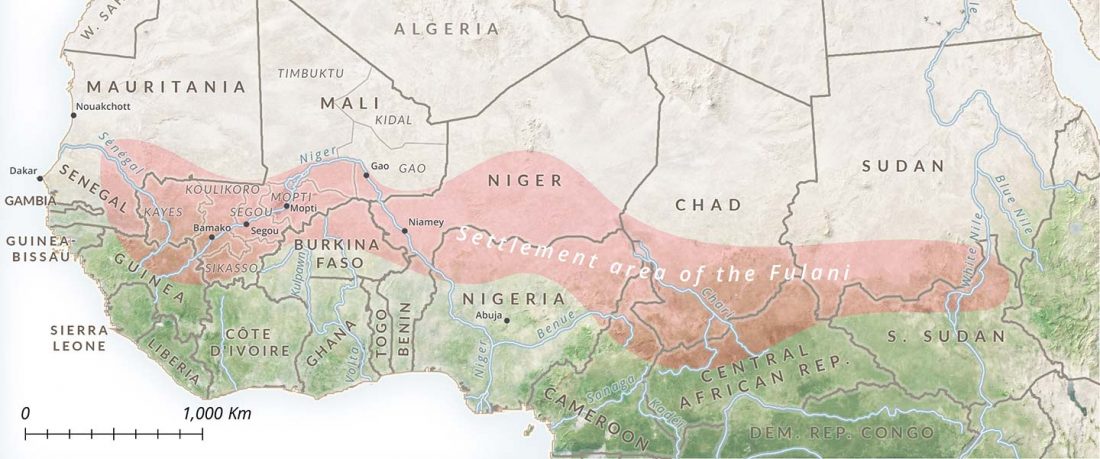
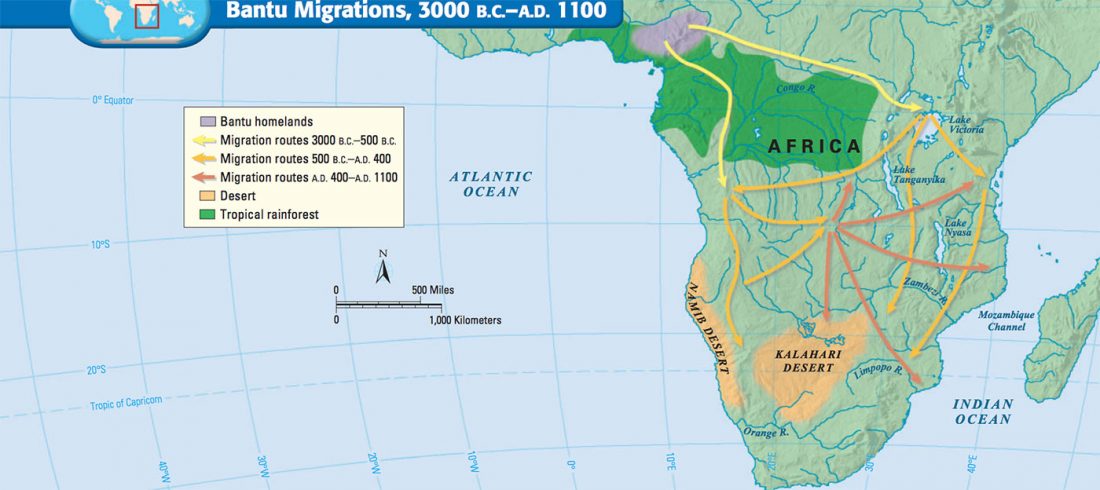
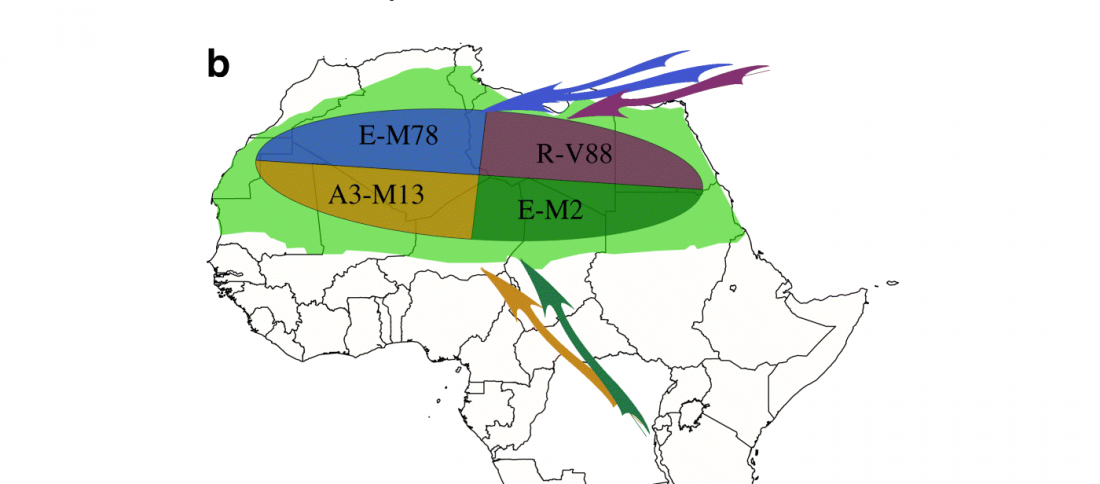
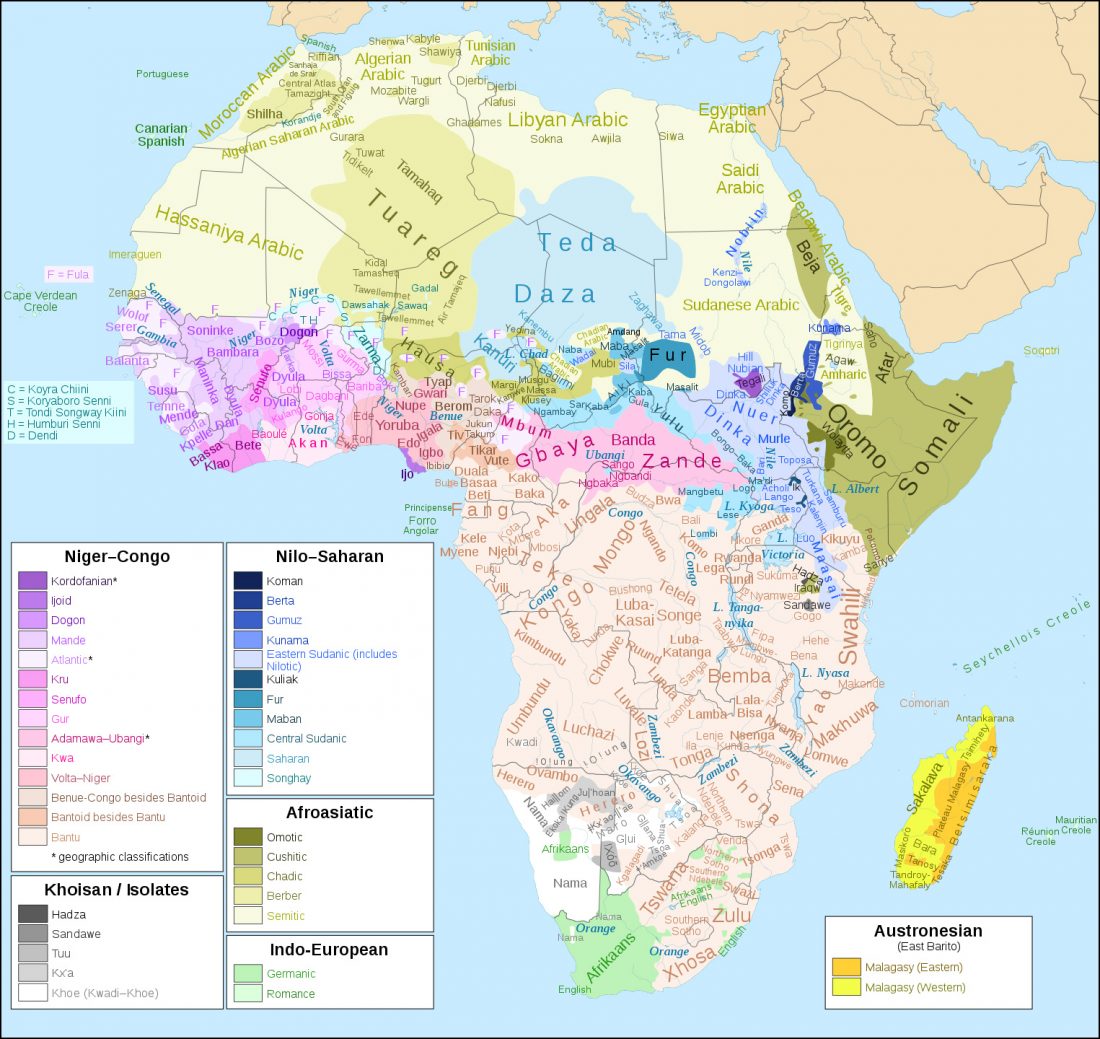
R1b in Eastern Arabia Late Neolithic / Bronze Age

A reader asked my opinion about my reported R1b subclade of one low quality sample from Ra’s Al-Ḥamrāʾ 5 necropolis, Muscat (Oman), published (without Y-DNA) in Harney et al. (2020). For those interested, here are the relevant calls, with information on the graves taken from Salvatori (2007):
I11919_I11920_I11921: Grave 221 (ca. 3700-3200 BC), mtDNA H2a2a1, Y-DNA R1bL754 (xPH155; xL389P297M269; xPF6323PF6292).
* The samples show a straightforward path (but full of deamination question marks): CT (with 1 ancestral call M5813 1x C-A) -PP295M45P284P226 -KM526YSC0000186 … Read the rest “R1b in Eastern Arabia Late Neolithic / Bronze Age”