Two interesting papers questioning previous methods have been published.
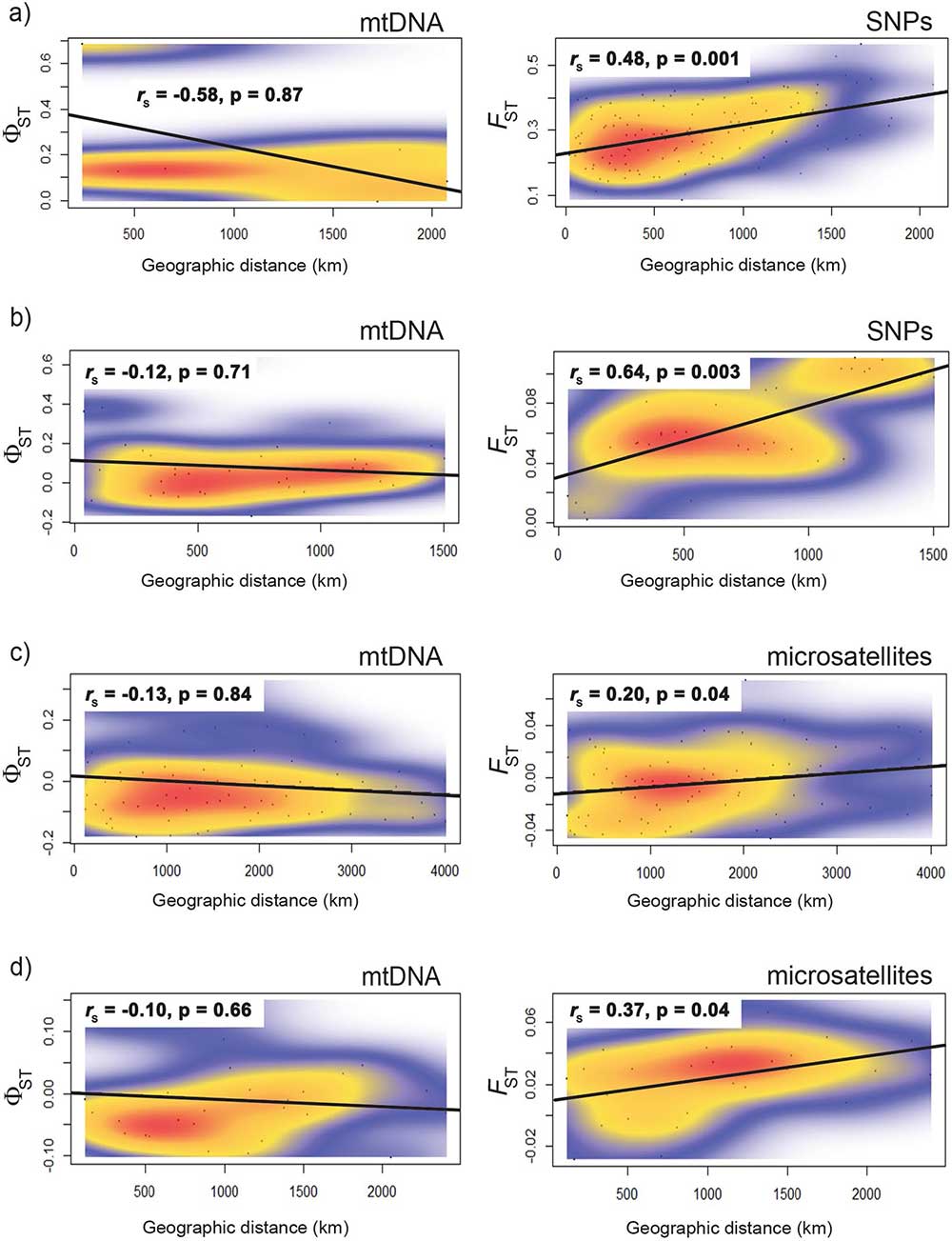
Open access Mitochondrial DNA is unsuitable to test for isolation by distance, by Teske et al. Scientific Reports (2018) 8:8448.
Abstract (emphasis mine):
Tests for isolation by distance (IBD) are the most commonly used method of assessing spatial genetic structure. Many studies have exclusively used mitochondrial DNA (mtDNA) sequences to test for IBD, but this marker is often in conflict with multilocus markers. Here, we report a review of the literature on IBD, with the aims of determining (a) whether significant IBD is primarily a result of lumping spatially discrete populations, and (b) whether microsatellite datasets are more likely to detect IBD when mtDNA does not. We also provide empirical data from four species in which mtDNA failed to detect IBD by comparing these with microsatellite and SNP data. Our results confirm that IBD is mostly found when distinct regional populations are pooled, and this trend disappears when each is analysed separately. Discrepancies between markers were found in almost half of the studies reviewed, and microsatellites were more likely to detect IBD when mtDNA did not. Our empirical data rejected the lack of IBD in the four species studied, and support for IBD was particularly strong for the SNP data. We conclude that mtDNA sequence data are often not suitable to test for IBD, and can be misleading about species’ true dispersal potential. The observed failure of mtDNA to reliably detect IBD, in addition to being a single-locus marker, is likely a result of a selection-driven reduction in genetic diversity obscuring spatial genetic differentiation.
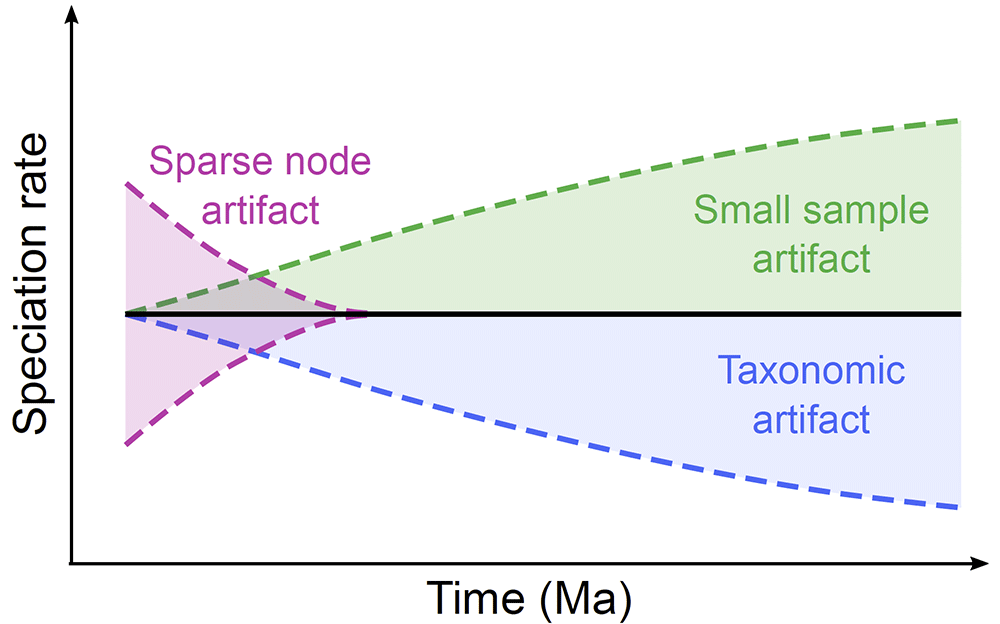
Behind paywall, Undersampling Genomes has Biased Time and Rate Estimates Throughout the Tree of Life, by Julie Marin and S. Blair Hedges, Mol Biol Evol (2018), msy103.
Abstract (emphasis mine):
Genomic data drive evolutionary research on the relationships and timescale of life but the genomes of most species remain poorly sampled. Phylogenetic trees can be reconstructed reliably using small data sets and the same has been assumed for the estimation of divergence time with molecular clocks. However, we show here that undersampling of molecular data results in a bias expressed as disproportionately shorter branch lengths and underestimated divergence times in the youngest nodes and branches, termed the small sample artifact. In turn, this leads to increasing speciation and diversification rates towards the present. Any evolutionary analyses derived from these biased branch lengths and speciation rates will be similarly biased. The widely used timetrees of the major species-rich studies of amphibians, birds, mammals, and squamate reptiles are all data-poor and show upswings in diversification rate, suggesting that their results were biased by undersampling. Our results show that greater sampling of genomes is needed for accurate time and rate estimation, which are basic data used in ecological and evolutionary research.
Do you remember the male-biased expansion from the Pontic-Caspian steppe, that was later contested in its methods by Lazaridis and Reich, but that is today again accepted by Reich and Lazaridis (probably for different reasons, namely Y-DNA evidence)?
Every time I read this kind of studies rejecting previous methods – which get written and published only because there is a future interest in them, not because they are (or may cause) retractions of previous results and interpretations – I remember these people inventing migration models based on genomic studies and saying “genetics is a science, linguistics/archaeology/anthropology is not”…
NOTE. Even if papers eventually receive a correction, journalists and blogs will keep echoing whatever gets published (see the famous Dennis/Denise will become dentists); there is no end to that. Believe it or not, we still see Underhill et al. (2014) being cited against the most recent papers, and even against the author’s own rejection of his paper’s results…
Especially right now, it must cause some kind of dissociated reasoning among those naysayers, when they need to resort to anthropological disciplines to discuss the latest interpretations of a potential Caucasus origin or North Iranian homeland of Proto-Indo-European…
EDIT (5 JUN 2018): Also, check out the recent review From genome-wide associations to candidate causal variants by statistical fine-mapping, by Schaid, Chen, and Larson.
Related:
- The Caucasus a genetic and cultural barrier; Yamna dominated by R1b-M269; Yamna settlers in Hungary cluster with Yamna
- Proto-Indo-European homeland south of the Caucasus?
- David Reich on social inequality and Yamna expansion with few Y-DNA subclades
- Post-Neolithic Y-chromosome bottleneck explained by cultural hitchhiking and competition between patrilineal clans
- Lazaridis’ evolutionary history of human populations in Europe
- Massive Migrations? The Impact of Recent aDNA Studies on our View of Third Millennium Europe
- Science and Archaeology (Humanities): collaboration or confrontation?