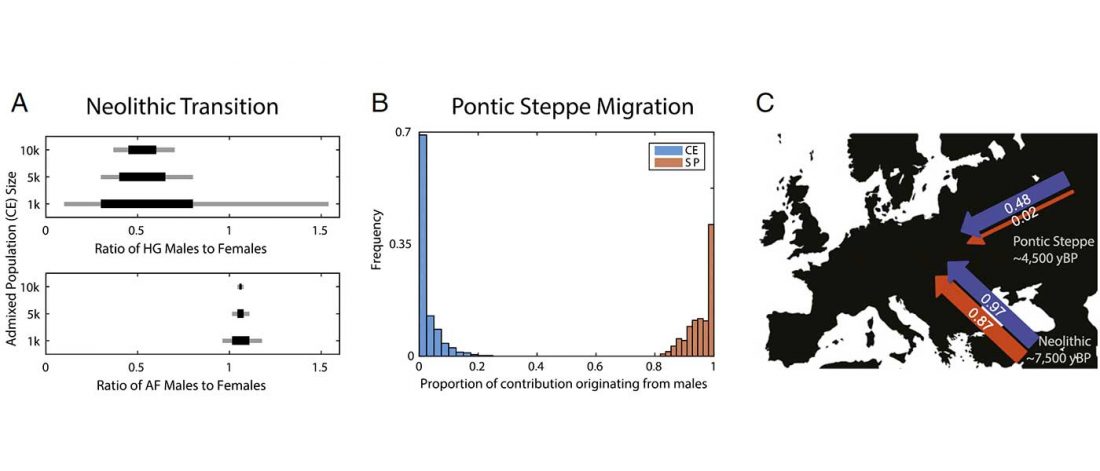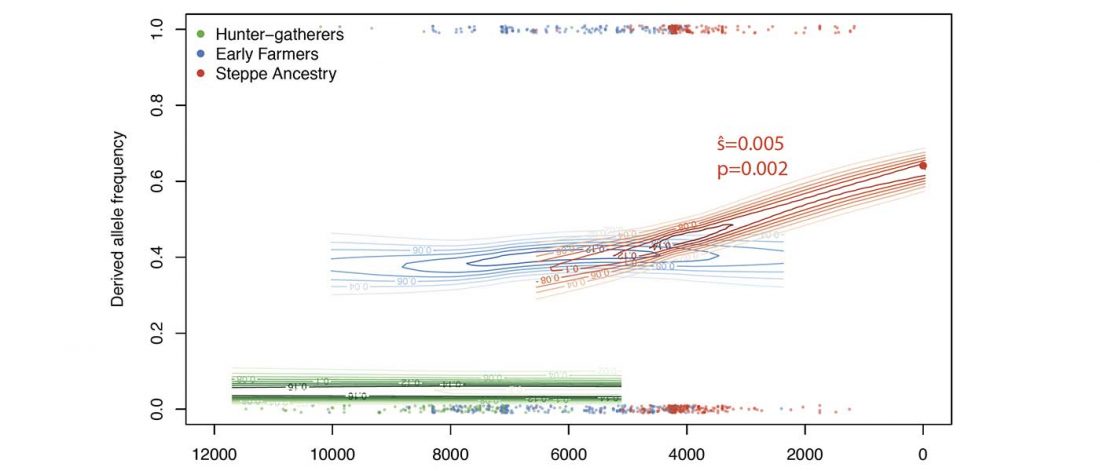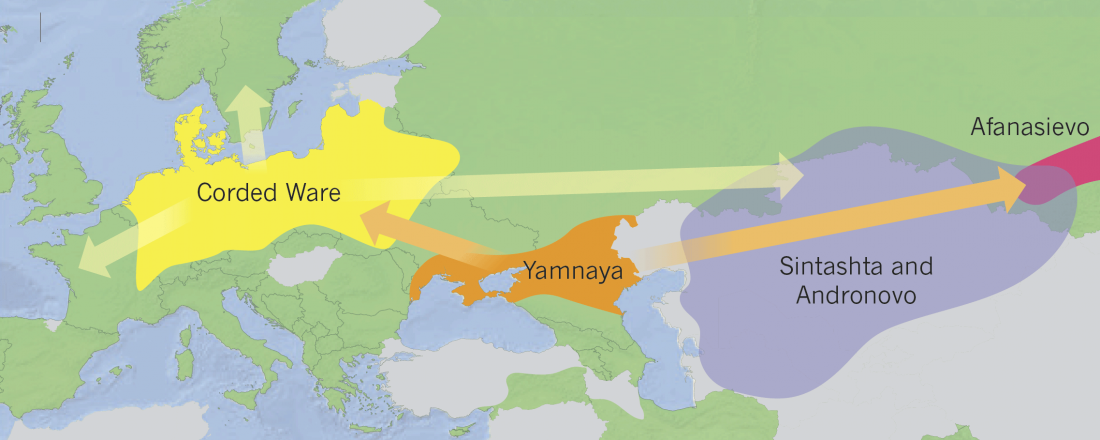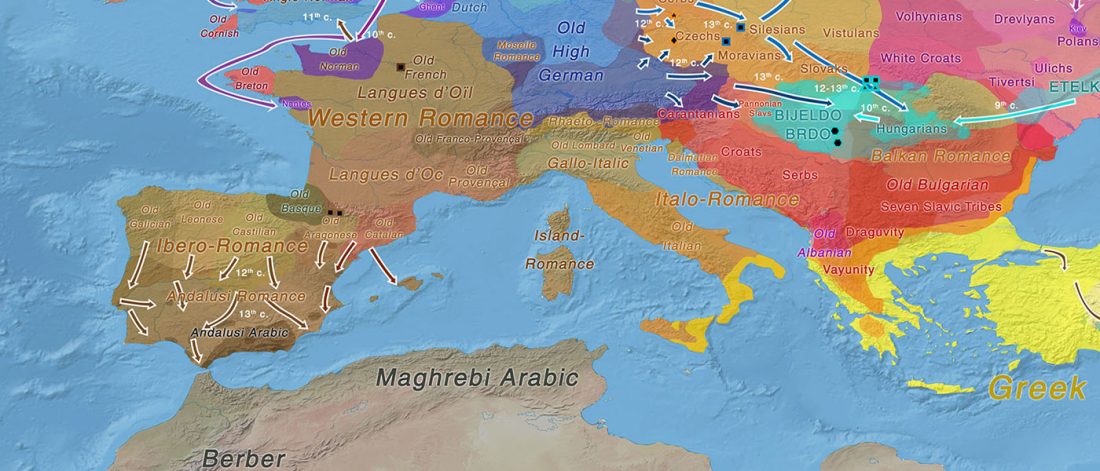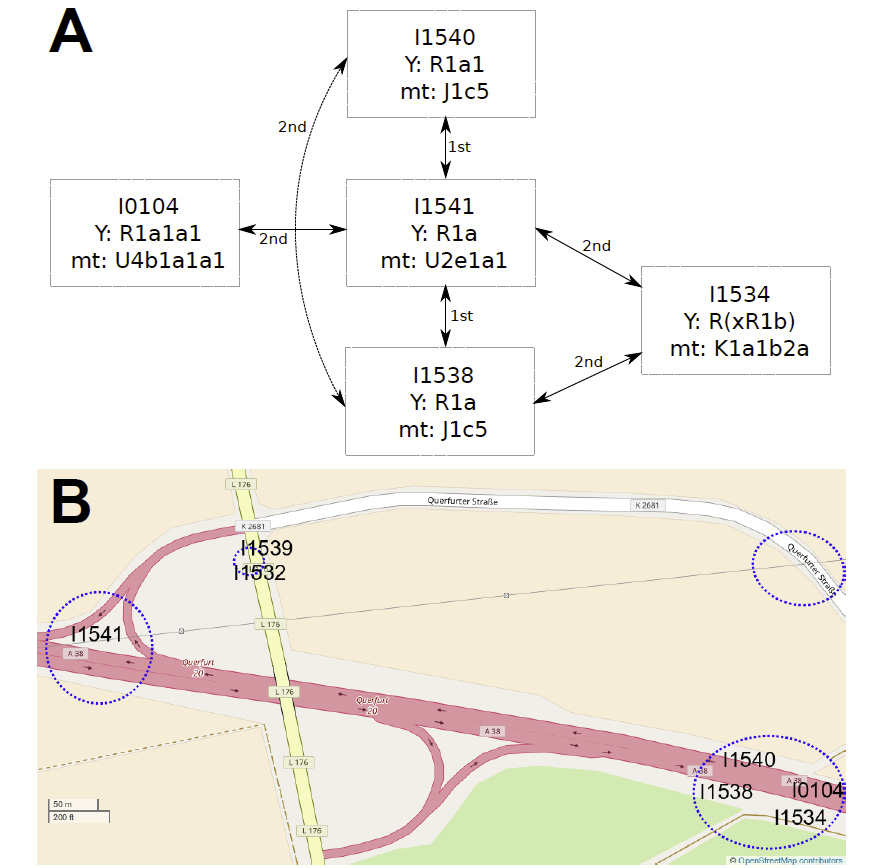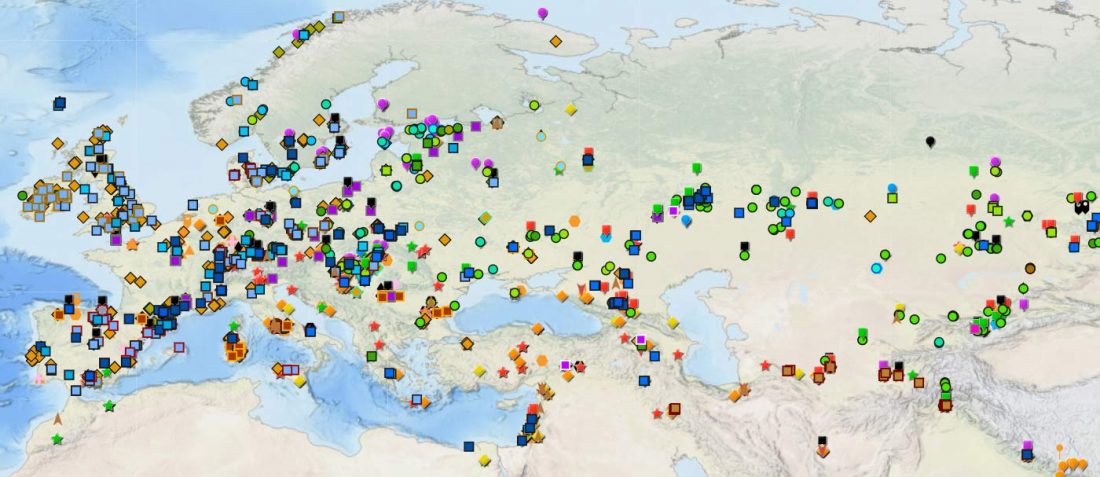
The last few weeks have been very exciting in terms the amount, diversity and quality of newly reported ancient samples, which included new genotypes and also Y-DNA and mtDNA haplogroups.
As some of you already know, I had been preparing a tailored GIS map of ancient DNA using QGIS-server on Ubuntu and trying some of the available plugins for the task, and was ready to use my old broken PC as a web server. For that, I needed to prepare different files corresponding to the different conventional divisions of the Prehistory Atlas. The crazy number of recently reported papers … Read the rest “Online GIS maps of ancient Y-DNA, mtDNA and ADMIXTURE”