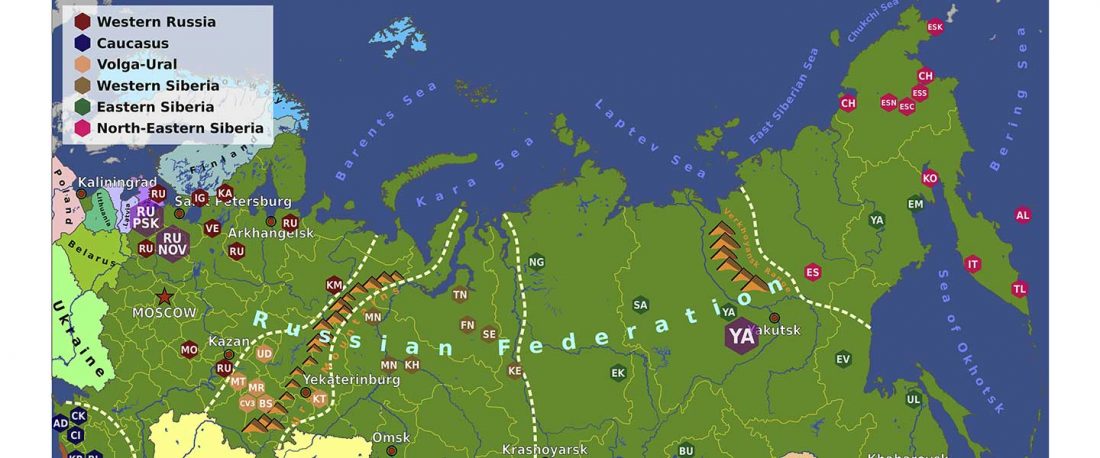
First look of an accepted manuscript (behind paywall), Genome-wide sequence analyses of ethnic populations across Russia, by Zhernakova et al. Genomics (2019).
Interesting excerpts:
… Read the rest “The cradle of Russians, an obvious Finno-Volgaic genetic hotspot”There remain ongoing discussions about the origins of the ethnic Russian population. The ancestors of ethnic Russians were among the Slavic tribes that separated from the early Indo-European Group, which included ancestors of modern Slavic, Germanic and Baltic speakers, who appeared in the northeastern part of Europe ca. 1,500 years ago. Slavs were found in the central part of Eastern Europe, where they came in direct contact with (and likely assimilation of) the populations speaking