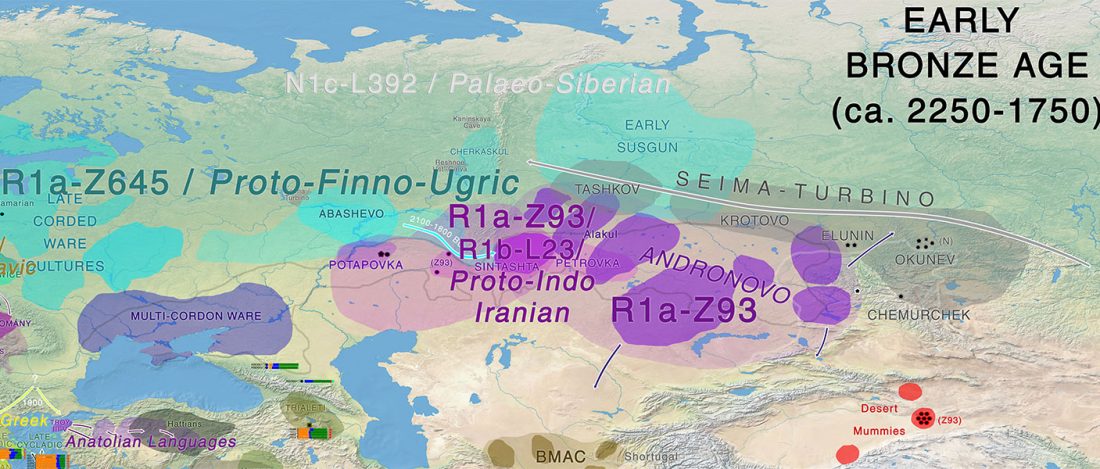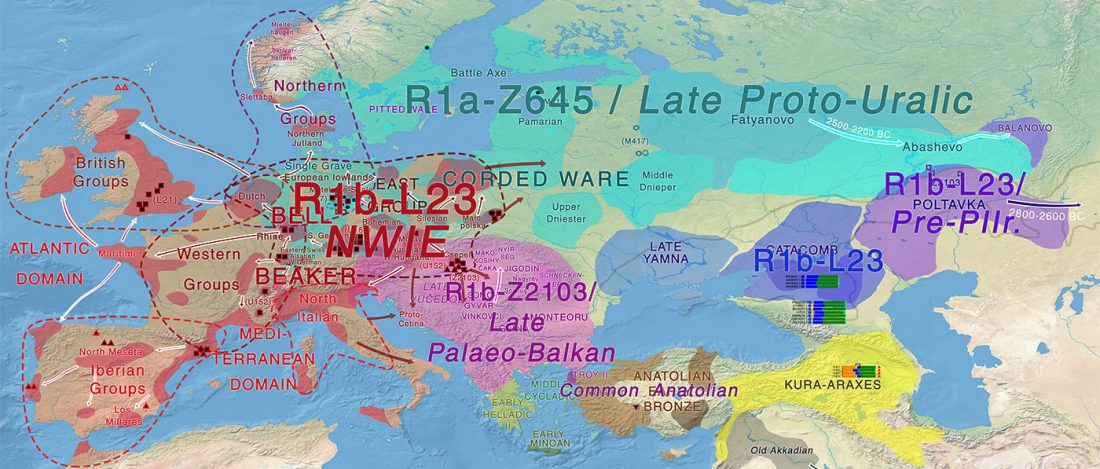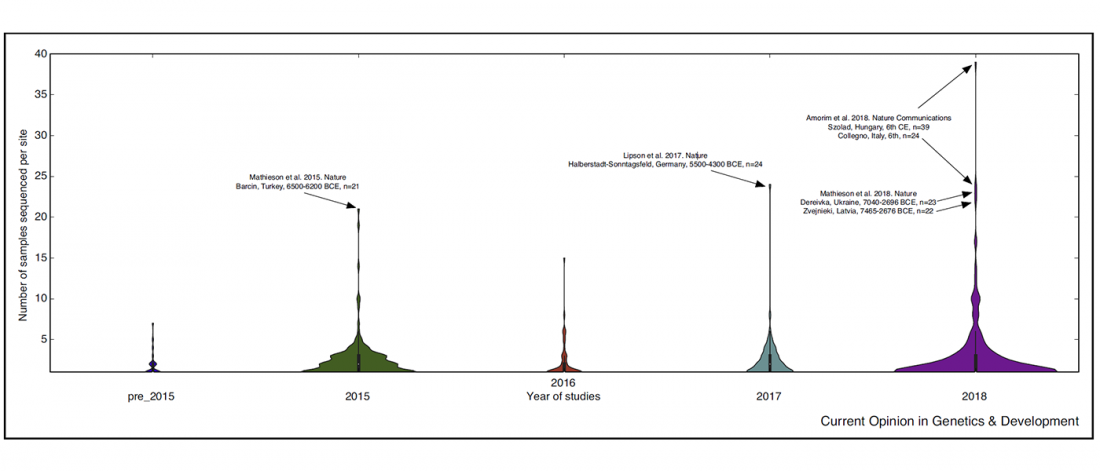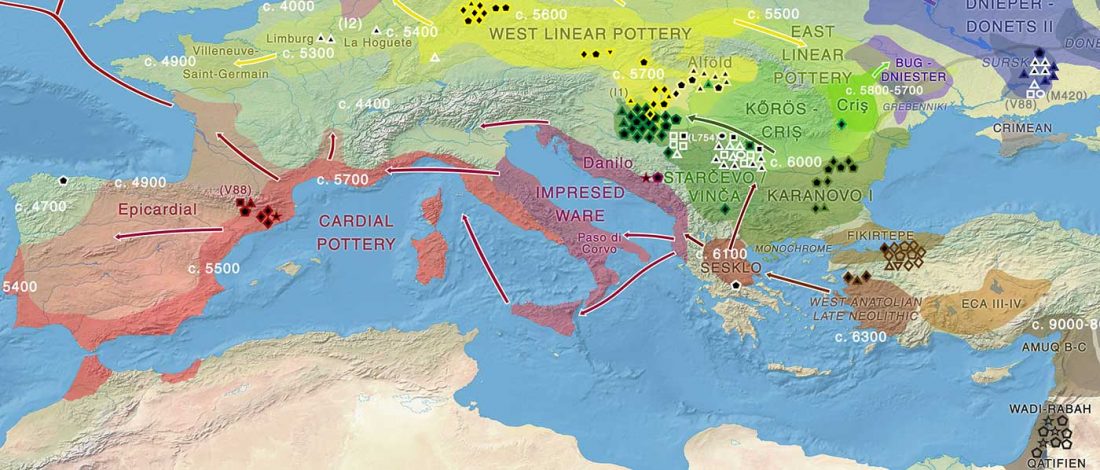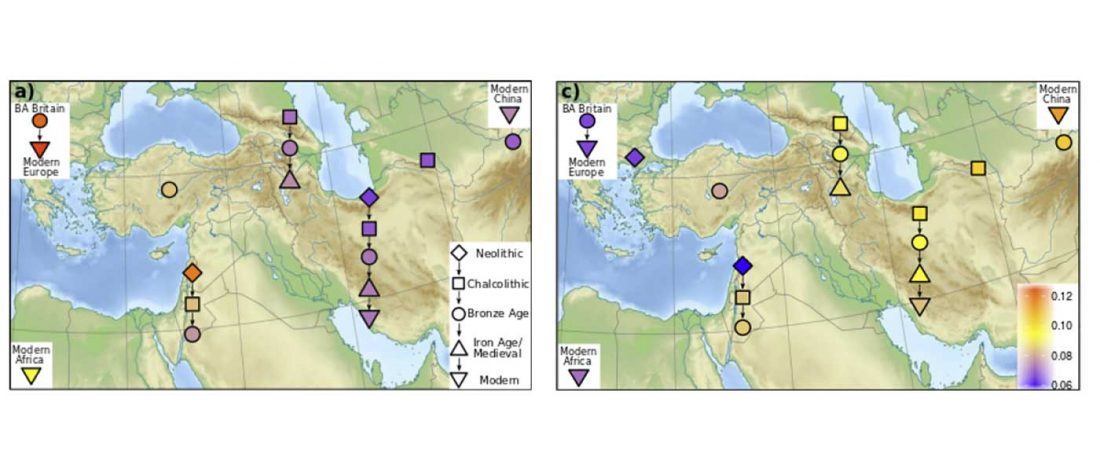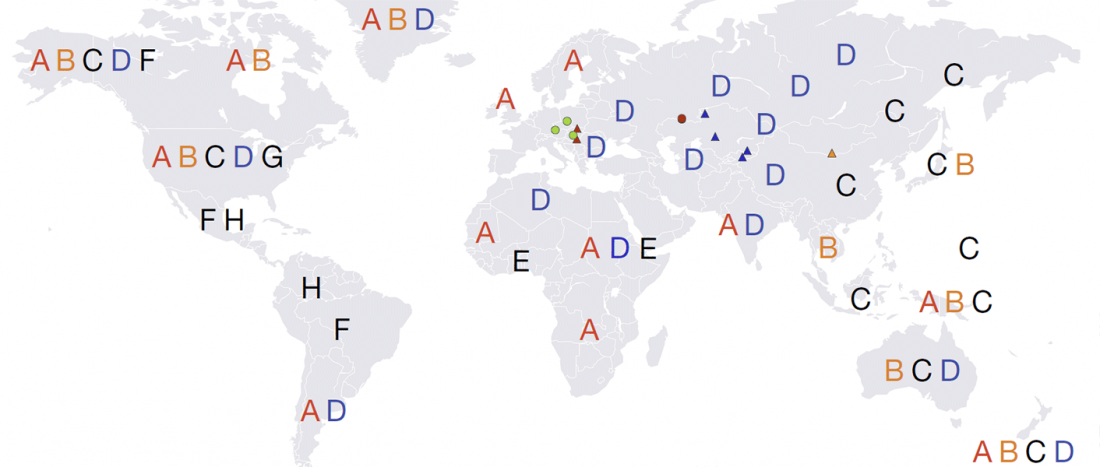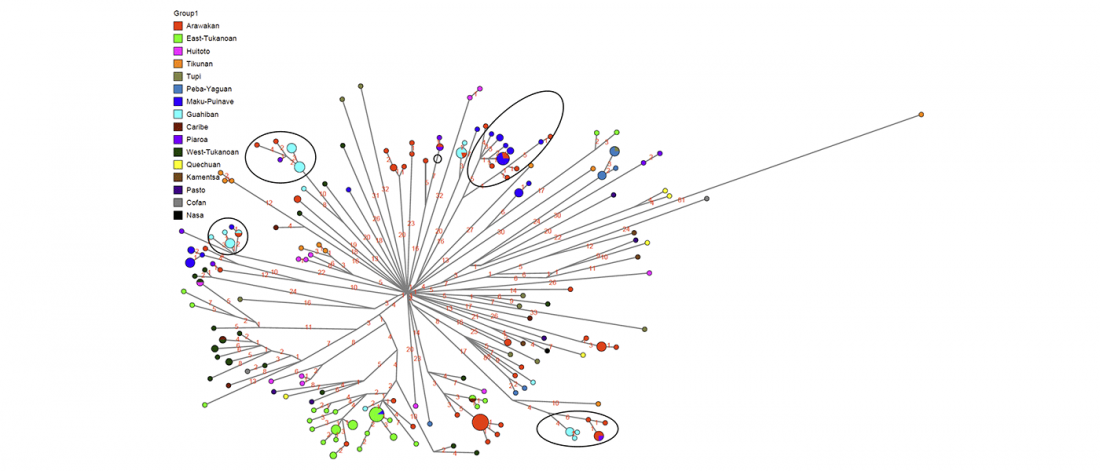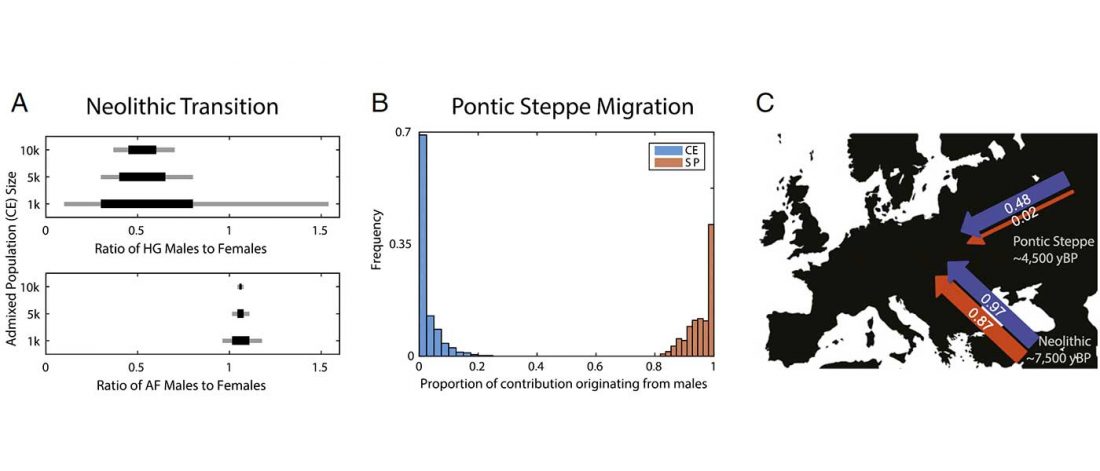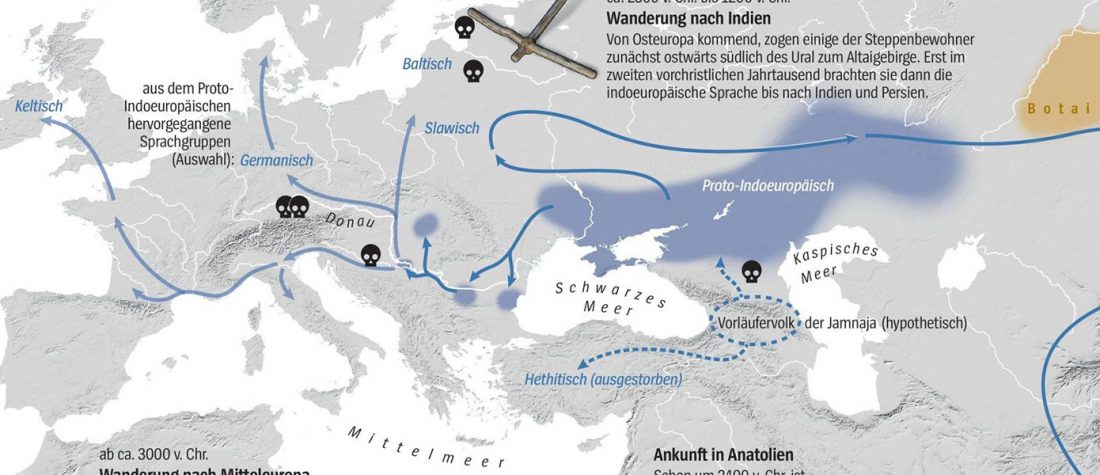
Good timing for the publication of two interesting papers, that a lot of people should read very carefully:
ADMIXTURE
Open access A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots, by Daniel J. Lawson, Lucy van Dorp & Daniel Falush, Nature Communications (2018).
Interesting excerpts (emphasis mine):
… Read the rest “Common pitfalls in human genomics and bioinformatics: ADMIXTURE, PCA, and the ‘Yamnaya’ ancestral component”Experienced researchers, particularly those interested in population structure and historical inference, typically present STRUCTURE results alongside other methods that make different modelling assumptions. These include TreeMix, ADMIXTUREGRAPH, fineSTRUCTURE, GLOBETROTTER, f3 and D statistics, amongst many others. These models can be used both to probe whether assumptions of the model