Ancient hepatitis B viruses from the Bronze Age to the Medieval period, by Mühlemann et al., Science (2018) 557:418–423.
NOTE. You can read the PDF at Dalia Pokutta’s Academia.edu account.
Abstract (emphasis):
Hepatitis B virus (HBV) is a major cause of human hepatitis. There is considerable uncertainty about the timescale of its evolution and its association with humans. Here we present 12 full or partial ancient HBV genomes that are between approximately 0.8 and 4.5 thousand years old. The ancient sequences group either within or in a sister relationship with extant human or other ape HBV clades. Generally, the genome properties follow those of modern HBV. The root of the HBV tree is projected to between 8.6 and 20.9 thousand years ago, and we estimate a substitution rate of 8.04 × 10−6–1.51 × 10−5 nucleotide substitutions per site per year. In several cases, the geographical locations of the ancient genotypes do not match present-day distributions. Genotypes that today are typical of Africa and Asia, and a subgenotype from India, are shown to have an early Eurasian presence. The geographical and temporal patterns that we observe in ancient and modern HBV genotypes are compatible with well-documented human migrations during the Bronze and Iron Ages1,2. We provide evidence for the creation of HBV genotype A via recombination, and for a long-term association of modern HBV genotypes with humans, including the discovery of a human genotype that is now extinct. These data expose a complexity of HBV evolution that is not evident when considering modern sequences alone.

Interesting excerpts:
We find genotype A in south-western Russia by 4.3 ka (in samples RISE386 and RISE387) in individuals belonging to the Sintashta culture, and in a Hungarian sample (DA195) from the Scythian culture. The western Scythians are related to the Bronze Age cultures of western steppe populations2 and their shared ancestry suggests that the modern genotype A may descend from this ancient Eurasian diversity and not, as previously hypothesized, from African ancestors29,30. This is also consistent with the phylogeny (Fig. 2), as well as the fact that the three oldest ancient genotype A sequences (HBV-DA195, HBV-RISE386 and HBV-RISE387) lack the six-nucleotide insertion found in the youngest (HBV-DA119) and in all modern genotype A sequences. The ancestors of subgenotypes A1 and A3 could have been carried into Africa subsequently, via migration from western Eurasia31.
The ancient HBV genotype D sequences were all found in Central Asia. HBV-DA27, found in Kazakhstan and dated to 1.6 ka, falls basal to the modern subgenotype D5 sequences that today are found in the Paharia tribe from eastern India32. DA27 and the Paharia people in India are linked by their East Asian ancestry2,33.
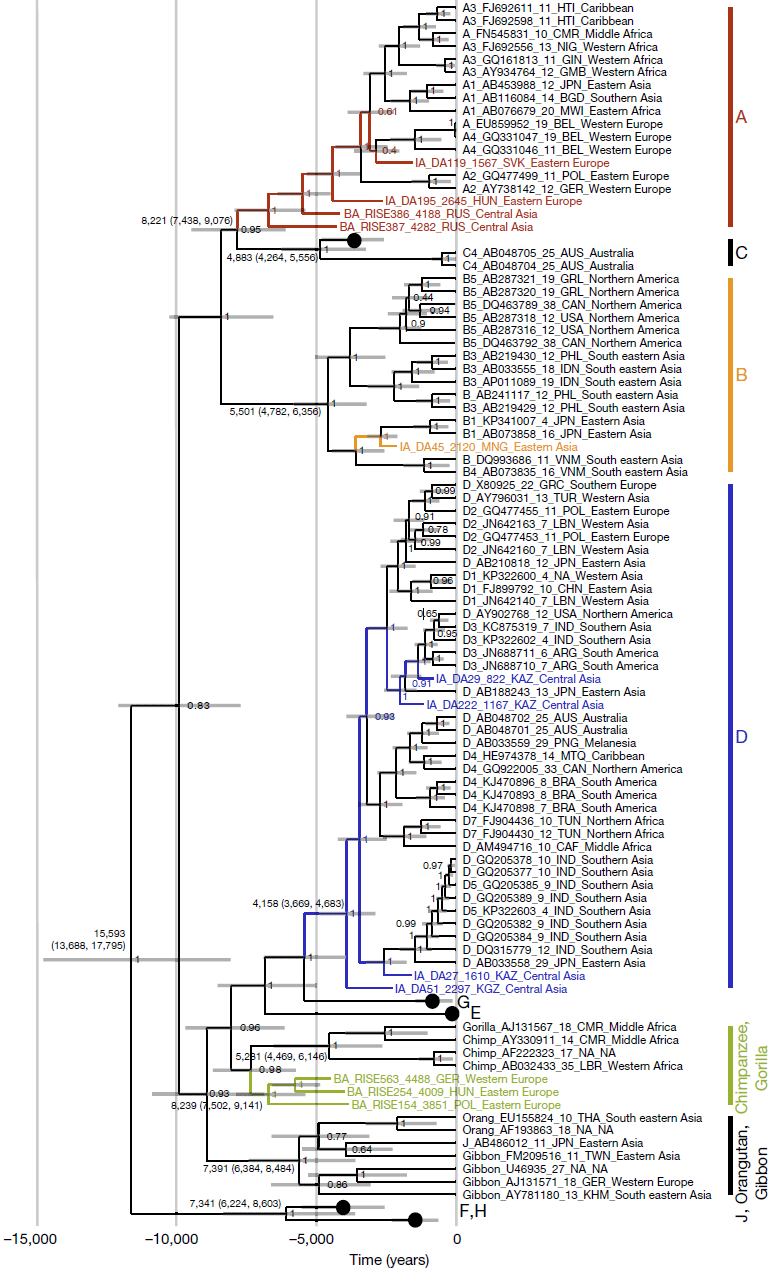
(…)Despite the age of the samples and the imperfect diagnostic test, our dataset contained a high proportion of HBV-positive individuals. The actual ancient prevalence during the Bronze Age and thereafter might have been higher, reaching or exceeding the prevalence typically found in contemporary indigenous populations5. This clearly establishes the potential of HBV as powerful proxy tool for research into human spread and interactions. The data from ancient genomes reveal aspects of complexity in HBV evolution that are not apparent when only modern sequences are considered. They show the existence of ancient HBV genotypes in locations incongruent with their present-day distribution, contradicting previously suggested geographical or temporal origins of genotypes or sub-genotypes; evidence for the creation of genotype A via recombination and the emergence of the genotype outside Africa; at least one now-extinct human genotype; ancient genotype-level localized diversity; and demonstrate that the viral substitution rate obtained from modern heterochronously sampled sequences is probably misleading. Together, these findings suggest that the difficulty in formulating a coherent theory for the origin and spread of HBV may be due to genetic evidence of an earlier evolutionary scenario being overwritten by relatively recent alterations, as has previously been suggested in the context of recombination24
See also:
- Oldest bubonic plague genome decoded in Srubna ca. 3800 YBP
- Phylogeny of leprosy, relevant for prehistoric Eurasian contacts
- Ancient DNA study reveals HLA susceptibility locus for leprosy in medieval Europeans
- Stone Age plague accompanying migrants from the steppe, probably Yamna, Balkan EBA, and Bell Beaker, not Corded Ware