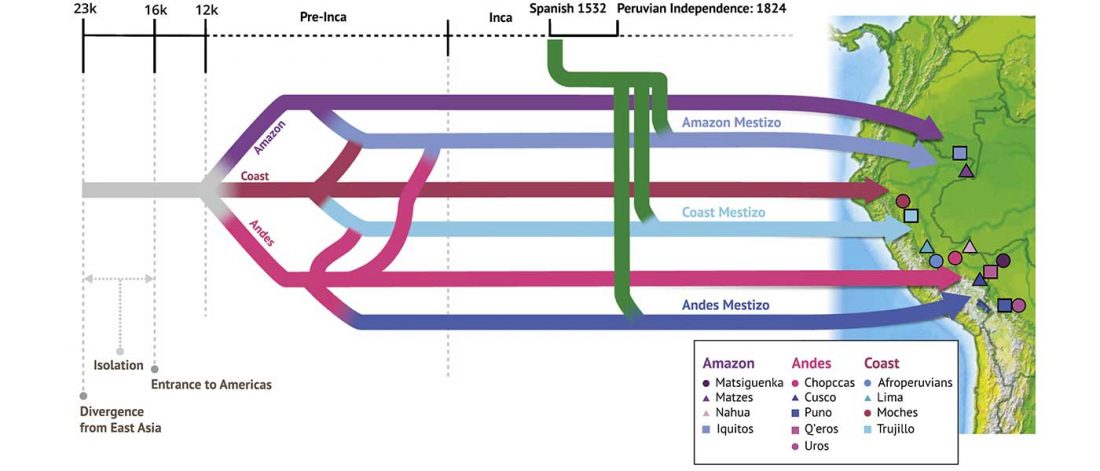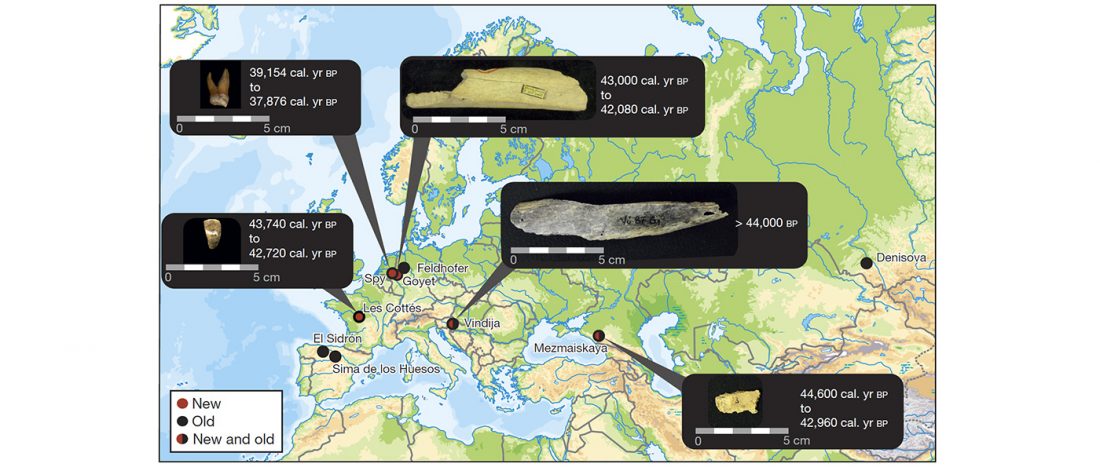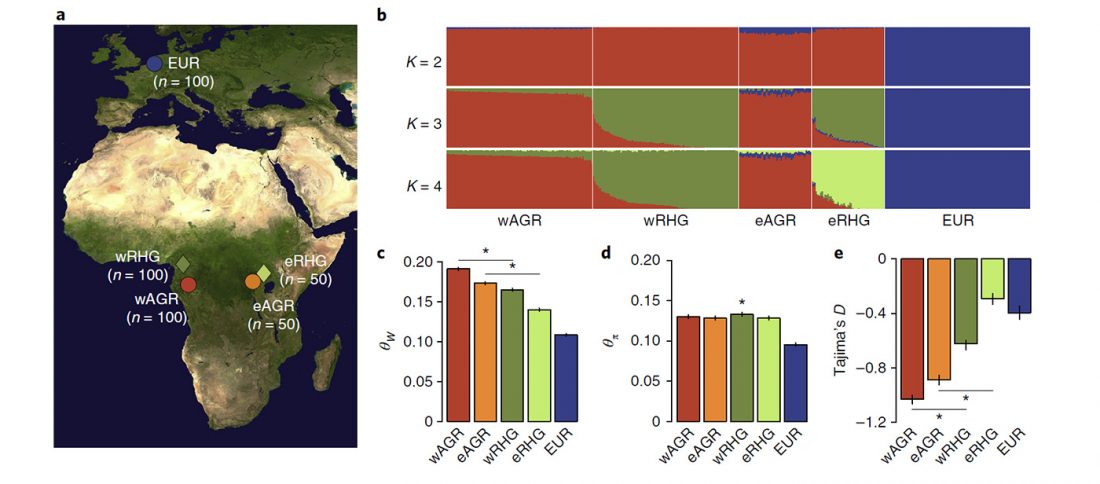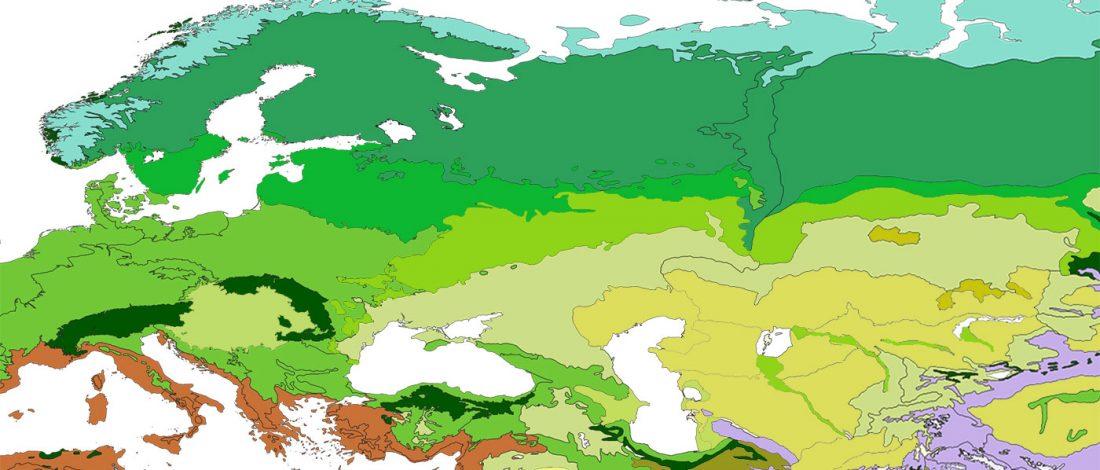
We know that the Caucasus Mountains formed a persistent prehistoric barrier to cultural and population movements. Nevertheless, an even more persistent frontier to population movements in Europe, especially since the Neolithic, is the Pontic-Caspian steppe – forest-steppe ecotone.
Like the Caucasus, this barrier could certainly be crossed, and peoples and cultures could permeate in both directions, but there have been no massive migrations through it. The main connection between both regions (steppe vs. forest-steppe/forest zone) was probably through its eastern part, through the Samara region in the Middle Volga.
The chances of population expansions crossing this natural … Read the rest “The genetic and cultural barrier of the Pontic-Caspian steppe – forest-steppe ecotone”