New paper (behind paywall) Paleolithic to Bronze Age Siberians Reveal Connections with First Americans and across Eurasia, by Yu et al. Cell (2020)
Interesting excerpts (emphasis mine, paragraphs subdivided for clarity):
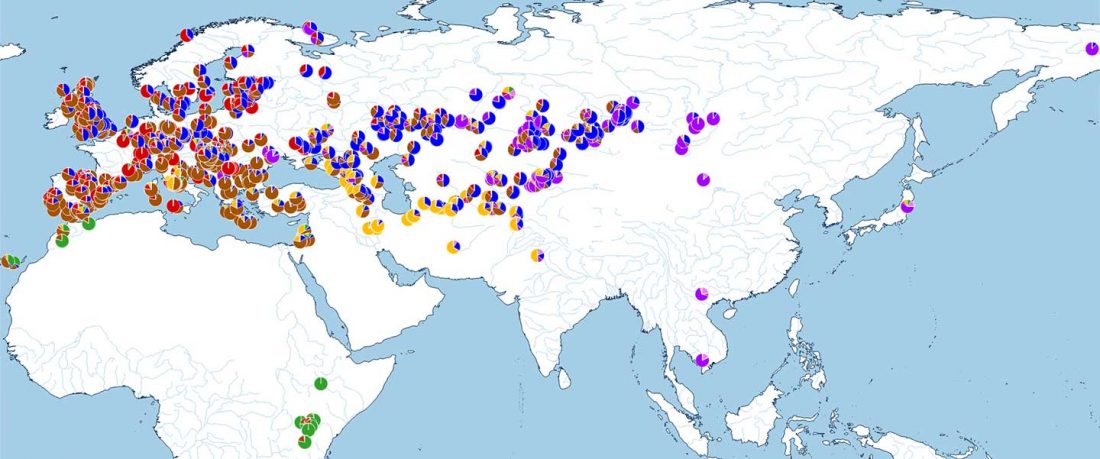
Population Structure (PCA)
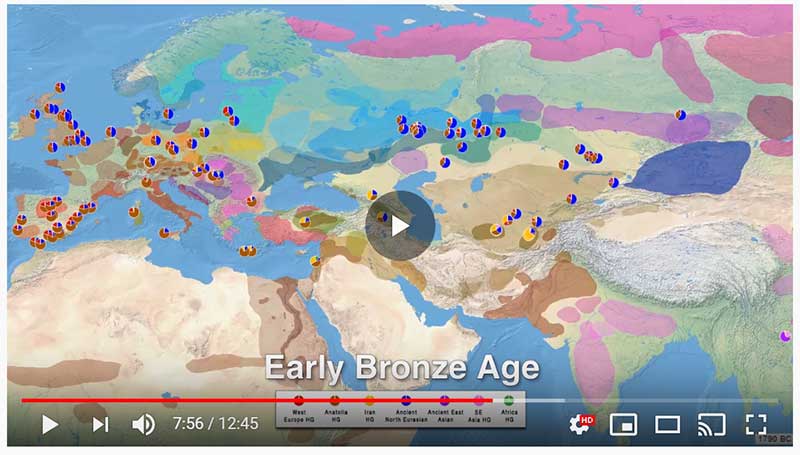
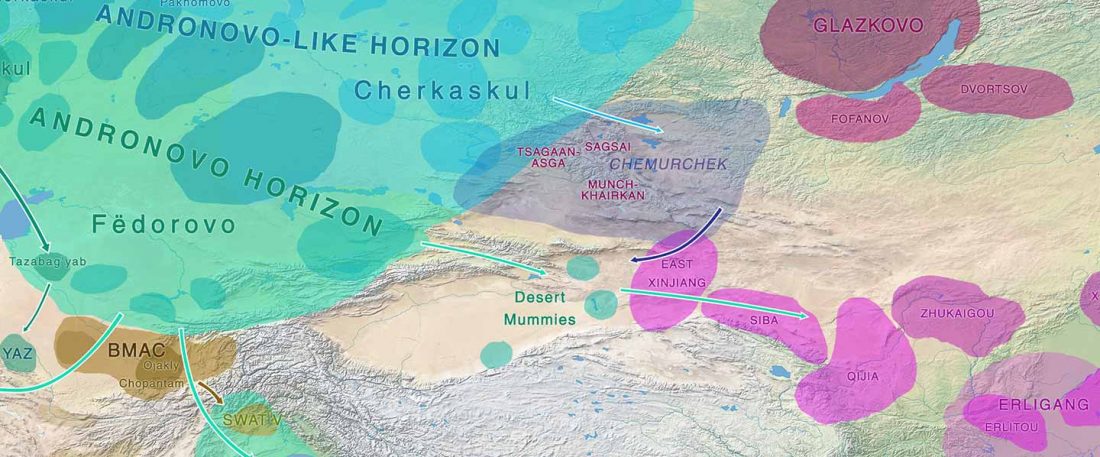
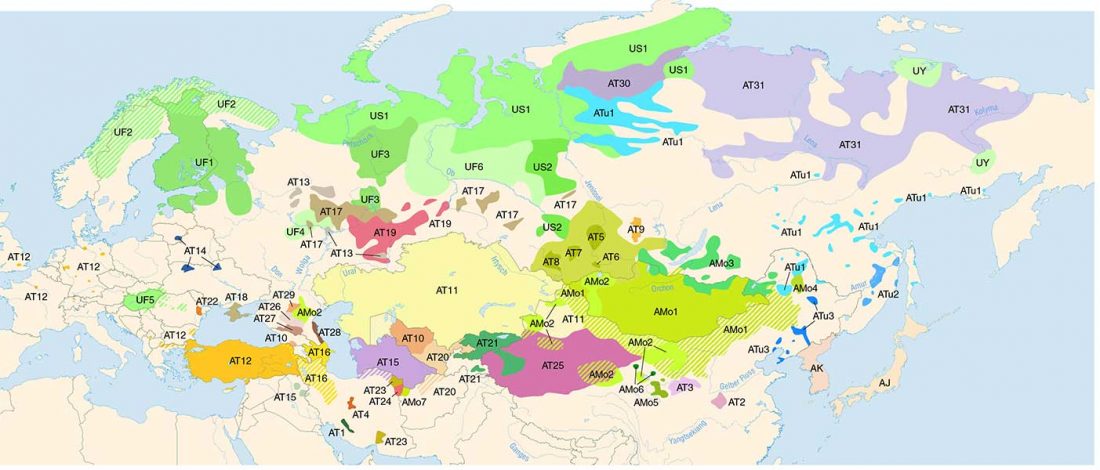
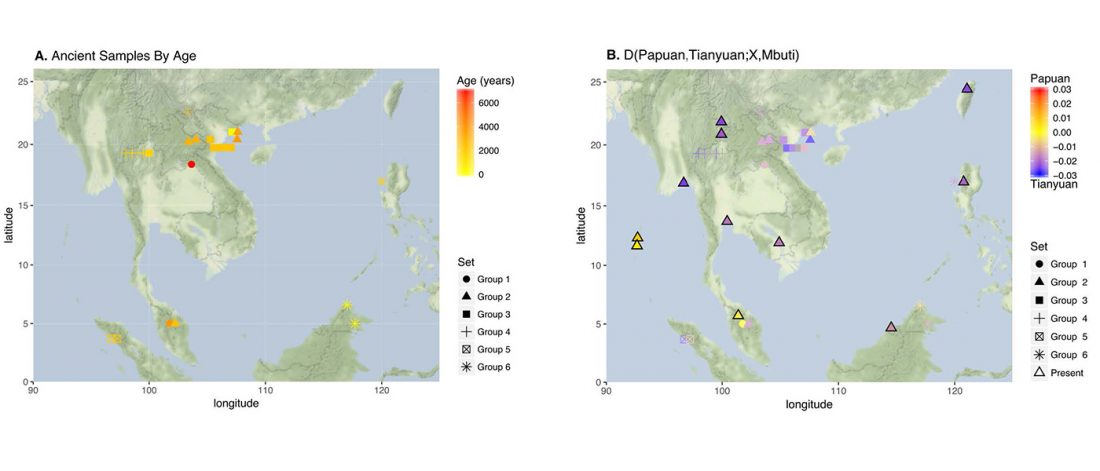
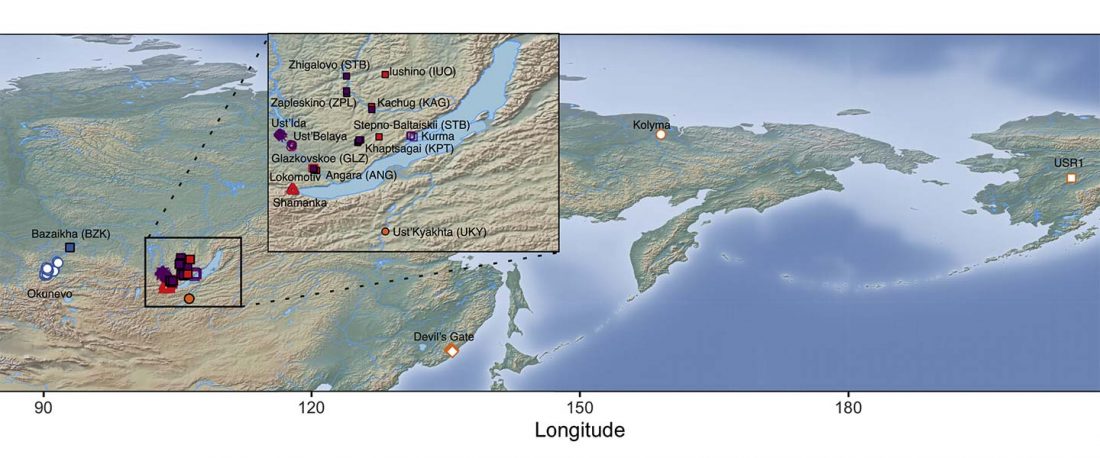
… Read the rest “Afanasievo ancestry reached Lake Baikal; Nganasan ancestry origins still at large”Most of the Lake Baikal individuals occupied the space on a “ANE-NEA” cline running between “Northeast Asian” (NEA) ancestry represented by Neolithic hunter-gathers from the Devil’s Gate in the Russian Far East (Sikora et al., 2019, Siska et al., 2017), and the ANE ancestry represented by Upper Paleolithic Siberian individuals MA1, AfontovaGora 2 (AG2), and AfontovaGora 3 (AG3) (Fu et al., 2016, Raghavan et al.,