Some interesting studies were published at roughly the same time as Damgaard et al. (Nature 2018 and Science 2018), and that’s probably why they got little attention (at least by me).
I’ve been disappointed that three recent exceptional studies of one of the world’s most historically important diseases, leprosy, have gotten so little notice from the science communication. It will take me a few hours to lay out their significance. But I think it’s important to do so.
So, here are the new studies on historical distribution and evolutionary development of Mycobacterium leprae, one of two organisms that causes leprosy (fourth study dropped yesterday!).
Leprosy is a chronic human disease caused by the yet-uncultured pathogen Mycobacterium leprae. Although readily curable with multidrug therapy (MDT), over 200,000 new cases are still reported annually. Here, we obtain M. leprae genome sequences from DNA extracted directly from patients’ skin biopsies using a customized protocol. Comparative and phylogenetic analysis of 154 genomes from 25 countries provides insight into evolution and antimicrobial resistance, uncovering lineages and phylogeographic trends, with the most ancestral strains linked to the Far East. In addition to known MDT-resistance mutations, we detect other mutations associated with antibiotic resistance, and retrace a potential stepwise emergence of extensive drug resistance in the pre-MDT era. Some of the previously undescribed mutations occur in genes that are apparently subject to positive selection, and two of these (ribD, fadD9) are restricted to drug-resistant strains. Finally, nonsense mutations in the nth excision repair gene are associated with greater sequence diversity and drug resistance.
Studying ancient DNA allows us to retrace the evolutionary history of human pathogens, such as Mycobacterium leprae, the main causative agent of leprosy. Leprosy is one of the oldest recorded and most stigmatizing diseases in human history. The disease was prevalent in Europe until the 16th century and is still endemic in many countries with over 200,000 new cases reported annually. Previous worldwide studies on modern and European medieval M. leprae genomes revealed that they cluster into several distinct branches of which two were present in medieval Northwestern Europe. In this study, we analyzed 10 new medieval M. leprae genomes including the so far oldest M. leprae genome from one of the earliest known cases of leprosy in the United Kingdom—a skeleton from the Great Chesterford cemetery with a calibrated age of 415–545 C.E. This dataset provides a genetic time transect of M. leprae diversity in Europe over the past 1500 years. We find M. leprae strains from four distinct branches to be present in the Early Medieval Period, and strains from three different branches were detected within a single cemetery from the High Medieval Period. Altogether these findings suggest a higher genetic diversity of M. leprae strains in medieval Europe at various time points than previously assumed. The resulting more complex picture of the past phylogeography of leprosy in Europe impacts current phylogeographical models of M. leprae dissemination. It suggests alternative models for the past spread of leprosy such as a wide spread prevalence of strains from different branches in Eurasia already in Antiquity or maybe even an origin in Western Eurasia. Furthermore, these results highlight how studying ancient M. leprae strains improves understanding the history of leprosy worldwide.
We report on a Mycobacterium leprae genome isolated from the remains of an individual with lepromatous leprosy that were excavated from a seventh‐century Hungarian cemetery. We determined that the genome was from a single nucleotide polymorphism (SNP) type 3K0 M. leprae strain, a lineage that diverged early from other M. leprae lineages. This is one of the earliest 3K0 M. leprae genomes to be sequenced to date. A number of novel SNPs as well as SNPs characteristic of the 3K0 lineage were confirmed by conventional polymerase chain reaction and Sanger sequencing. Recovery of accompanying human DNA from the burial was poor, particularly when compared with that of the pathogen. Modern 3K0 M. leprae strains have only been isolated from East Asia and the Pacific, and so these findings require new scenarios to describe the origins and routes of dissemination of leprosy during antiquity that have resulted in the modern phylogeographical distribution of M. leprae.
Leprosy is caused by the bacterial pathogens Mycobacterium leprae and Mycobacterium lepromatosis. Apart from humans, animals such as nine-banded armadillos in the Americas and red squirrels in the British Isles are naturally infected with M. leprae. Natural leprosy has also been reported in certain nonhuman primates, but it is not known whether these occurrences are due to incidental infections by human M. leprae strains or by M. leprae strains specific to nonhuman primates. In this study, complete M. leprae genomes from three naturally infected nonhuman primates (a chimpanzee from Sierra Leone, a sooty mangabey from West Africa, and a cynomolgus macaque from The Philippines) were sequenced. Phylogenetic analyses showed that the cynomolgus macaque M. leprae strain is most closely related to a human M. leprae strain from New Caledonia, whereas the chimpanzee and sooty mangabey M. leprae strains belong to a human M. leprae lineage commonly found in West Africa. Additionally, samples from ring-tailed lemurs from the Bezà Mahafaly Special Reserve, Madagascar, and chimpanzees from Ngogo, Kibale National Park, Uganda, were screened using quantitative PCR assays, to assess the prevalence of M. leprae in wild nonhuman primates. However, these samples did not show evidence of M. leprae infection. Overall, this study adds genomic data for nonhuman primate M. leprae strains to the existing M. leprae literature and finds that this pathogen can be transmitted from humans to nonhuman primates as well as between nonhuman primate species. While the prevalence of natural leprosy in nonhuman primates is likely low, nevertheless, future studies should continue to explore the prevalence of leprosy-causing pathogens in the wild.
These five studies are doing whole-genome sequencing on either modern isolates of M. leprae, or genomic fragments retrieved from buried remains (aDNA). The main objective of all the studies is to understand the diversity of M. leprae, both in terms of its history and in terms of its present-day distribution. (Benjak et al. 2018 are especially concerned to study possible reasons for variance in multiple drug resistance).
The following comments are concerned only to discuss leprosy’s history.
So, let’s start with a common claim of the science communication pieces on Schuenemann et al. 2018, which was published last week. A common formula: “New Study Suggests Leprosy Began To Spread From Europe To The World“. Is it plausible that Europe was where leprosy originated as human disease?
The answer, actually, is no. There’s two reasons for this, one having to do with chronology, the other with geography.
For chronology, these studies cumulatively suggest we are looking at a bottleneck. The current Time to Most Recent Common Ancestor (TMRCA) suggested for the divergence of M. leprae from its closest known “cousin,” M. lepromatosis (which also causes leprosy in humans) is estimated to be ca. 13.9 million years. There were no humans around 13.9M ya. So we cannot have been M. leprae‘s original host. All studies being discussed here agree on a consensus phylogeny, which puts the origin of all known strains of M. leprae at about 4-5K ya. So when we talk about the “origin” of M. leprae, we only talking about those lineages formed after this bottleneck.
Next we have to look at geography. Let’s start with this statement from the most recent study, Mendum et al. 2018, which is discussing a genome sequenced from an individual in Hungary from the 7th c. CE: “Modern 3K0 M. leprae strains have only been isolated from East Asia and the Pacific and so these findings require new scenarios to describe the origins and routes of dissemination of leprosy during antiquity that have resulted in the modern phylogeographical distribution of M. leprae.”
Okay, so stop and consider the implications of this. We have someone from 7th c. Hungary with leprosy. The strain of M. leprae that he has is not most closely related to strains sequenced earlier in Denmark or Sweden or England (see Schuenemann et al. 2018, refs. 9, 20, & 21). Rather, the strain he has (3K0) is most closely related to modern strains currently documented on the Pacific Rim, a very, very long way from Hungary. Here are the summary reflections of Mendum et al. 2018:
The global distribution of 3K0 and 3K1 strains is today restricted to regions of the Western Pacific such as Japan (except Okinawa), Korea, China, The Philippines, New Caledonia and Indonesia amongst others (Kai et al, 2013; Avanzi et al, 2015; Monot el al, 2009; Weng et al, 2013; Honap et al, 2018). This could indicate that the 3K lineage originated in Northern or Eastern Asia. The presence of two type 3K cases (KD271 and 222) in early medieval Hungary would then suggest a route of dissemination from Asia to central Europe, perhaps via trade links or migrations. This would be consistent with what is known of the origins of the Pannonian Avars, who are believed to have reached the Hungarian plain from the Eurasian steppe in the late 6th to early 9th centuries (Curta, 2006). The other possibility is that Europe was a centre of dissemination of the ancestral 3K0 and related strains, some of which later became less common or even absent from Europe but persisted in East Asia and the Pacific. Determining the likelihood of each of these scenarios will require more sampling and characterisation of both ancient and modern strains.
Two things bear stressing:
The lineage in which the Hungarian sample has been placed, Lineage 0, has now been documented in historical remains from Denmark, too. (Schuenemann et al. 2018) So whatever transmission routes are postulated to connect the Pacific Rim to Hungary, we will also need to postulate routes to connect the Pacific Rim to Denmark.
Schuenemann et al. 2018 document four of the five known M. leprae lineages in medieval western Europe. (Mendum et al. 2018 now declare the existence of 6 lineages; see tree.)
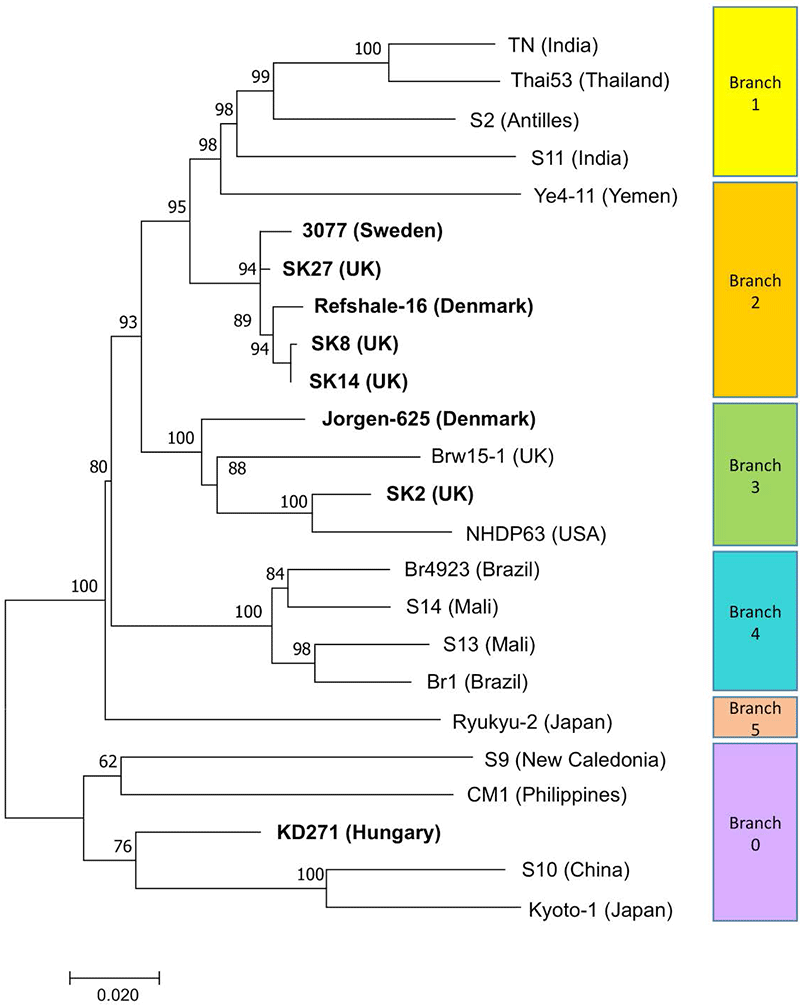
Phylogenetic relationships between selected modern (regular text) and ancient (bold text) M. leprae strains. The phylogeny was inferred by the Maximum Likelihood method of MEGA7 (Kumar et al, 2016) and the Tamura-3-Parameter model. The tree with the highest log likelihood value is shown. Bootstrap percentages from 1000 replicates are shown next to the branches. The scale indicates the number of substitutions per site. All positions with less than 90% site coverage were eliminated. M. lepromatosis was used as an outgroup (not shown). CM1 and Br15-1 are derived from a cynomolgus macaque and a red squirrel respectively.
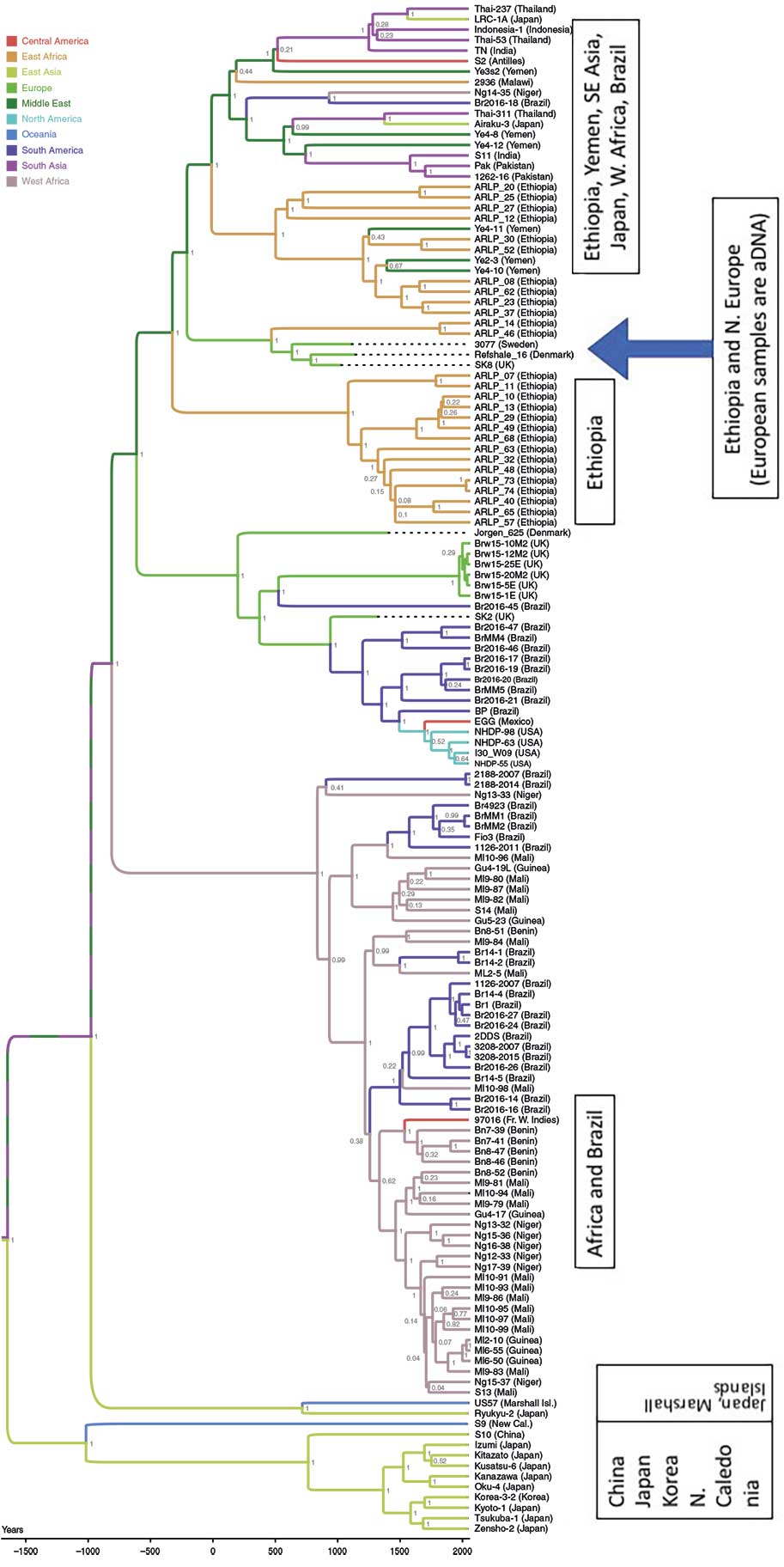
Now, remember that we also need to keep chronology in mind: Lineage 0 is thought to have diverged from the common ancestor of Lineages 1-4 at least 3.5K ya. (Here’s the phylogenetic tree from Benjak et al. 2018, which I have marked with time divisions for emphasis.)
Modified image by Monica H. Green. Phylogeny of M. leprae. Bayesian phylogenetic tree of 146 genomes of M. leprae calculated with BEAST 2.4.4. Hypermutated samples with mutations in the nth gene were excluded from the analysis. The tree is drawn to scale, with branch lengths representing years of age. Samples were binned according to geographic origin as given in the legend. Posterior probabilities for each node are shown in gray. Location probabilities of nodes were inferred by the Discrete Phylogeny model
So what we need to explain is how a strain (Lineage 0, or 3K0 as Mendum et al. 2018 call it) can be found all the way from Denmark to New Caledonia. An “Out of Europe” narrative isn’t really helpful, any more than the earlier “Out of Africa” narrative worked.
Given the extreme amount of suffering leprosy has caused, and continues to cause around the world, and given the extraordinary investigative power that paleogenetics has now developed, it’s really time that we did a better job pulling these global narratives together.
If you have Twitter, be sure to retweet this thread!
I've been disappointed that 3 recent exceptional studies of one of the world's most historically important diseases, #leprosy, have gotten so little notice from #scicomm. It will take me a few hours to lay out their significance. But I think it's important to do so. 1/n
Plague outbreaks in the past are mainly known from written sources; in particular, the Justinianic Plague of the Early Middle Ages and the Black Death of the Late Middle Ages have been described in vivid detail. Yet prior to the introduction of aDNA analysis, it was often quite difficult to associate burials with plague beyond doubt – especially in areas where written evidence of the plague is scarce. As analysis of ancient DNA now allows the detection of plague victims in the archaeological record, new ways are being developed for combining archaeological, historical and ancient DNA research. In this paper we would like to present and compare known examples of plague graves from the Early Middle Ages, the Late Middle Ages and the Thirty Years’ War in Germany that have also been confirmed by ancient DNA analyses. We would like to argue for a differentiated view of the burial customs, especially when more than one plague victim shared a grave, and would like to show possible conclusions, drawn from the aDNA-confirmed plague burials, that can indicate the different strategies adopted by ancient societies to deal with catastrophic events like a pandemic disease.
It is good practice to be registered
and logged in to comment.
Please keep the discussion of this post on topic. Civilized discussion. Academic tone.
For other topics, use the forums instead.