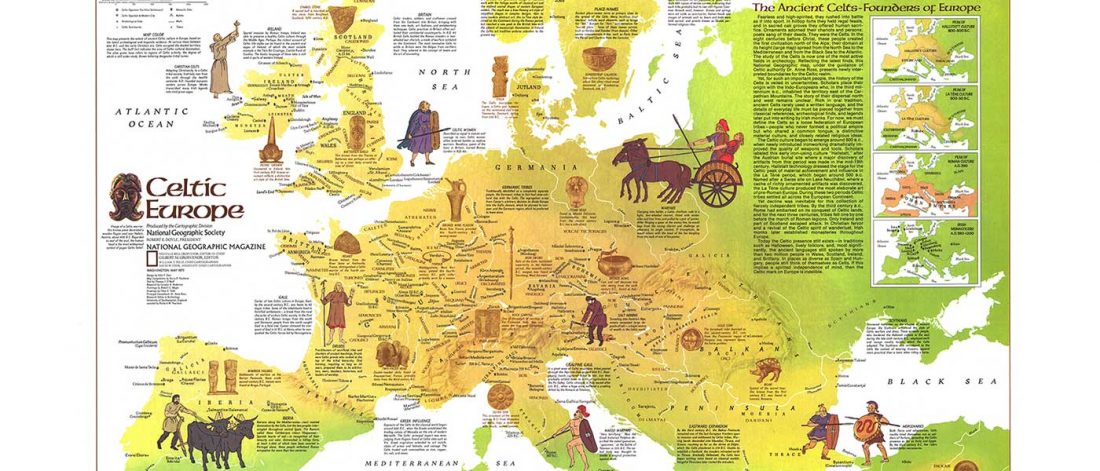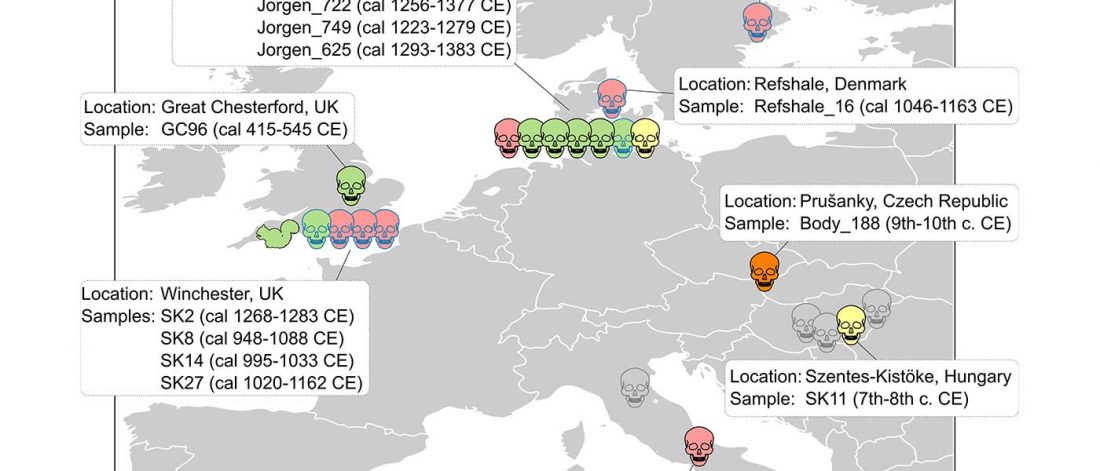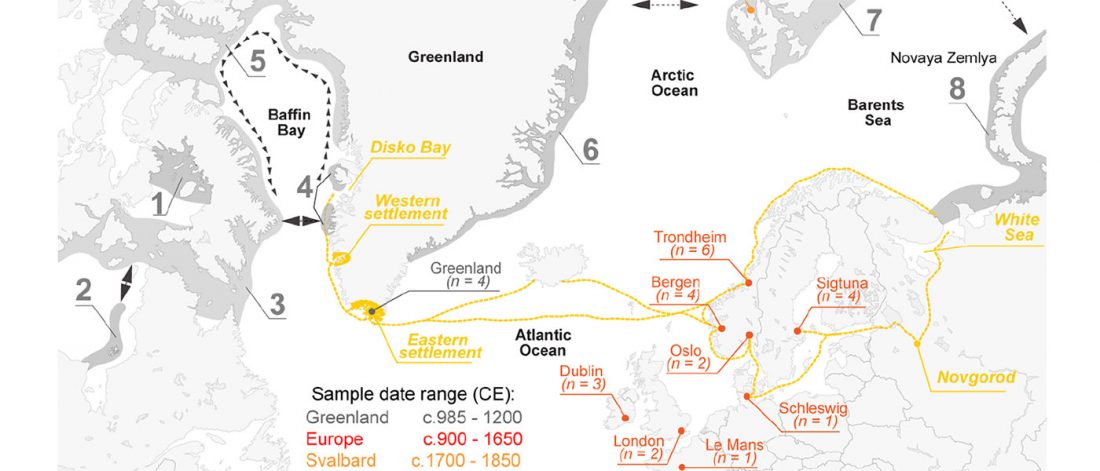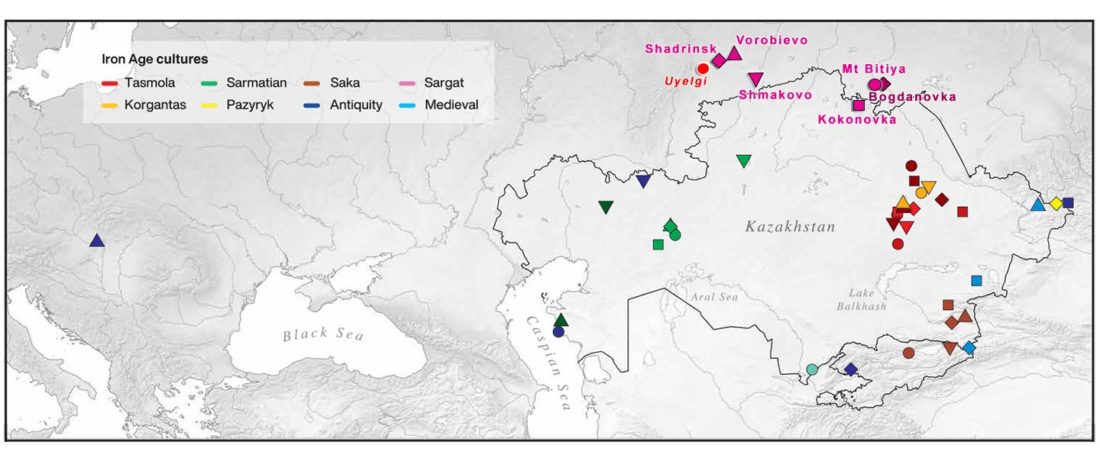
Open access Ancient genomic time transect from the Central Asian Steppe unravels the history of the Scythians, by Gnecchi-Ruscone et al. Sci Adv. 7 (13) eabe4414.
Interesting excerpts (emphasis mine):
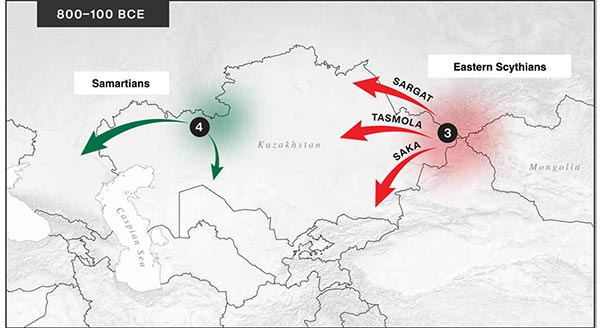
… Read the rest “Iron Age nomads of West Siberia of hg. Q1b, R1a, and basal N1a-L1026”From an archaeological perspective, the earliest IA burials associated with nomad-warrior cultures were identified in the eastern fringes of the Kazakh Steppe, in Tuva and the Altai region (ninth century BCE).
Following this early evidence, the Tasmola culture in central and north Kazakhstan is among the earliest major IA nomad warrior cultures emerging (eighth to sixth century BCE).
These earlier groups were followed by the iconic Saka cultures located