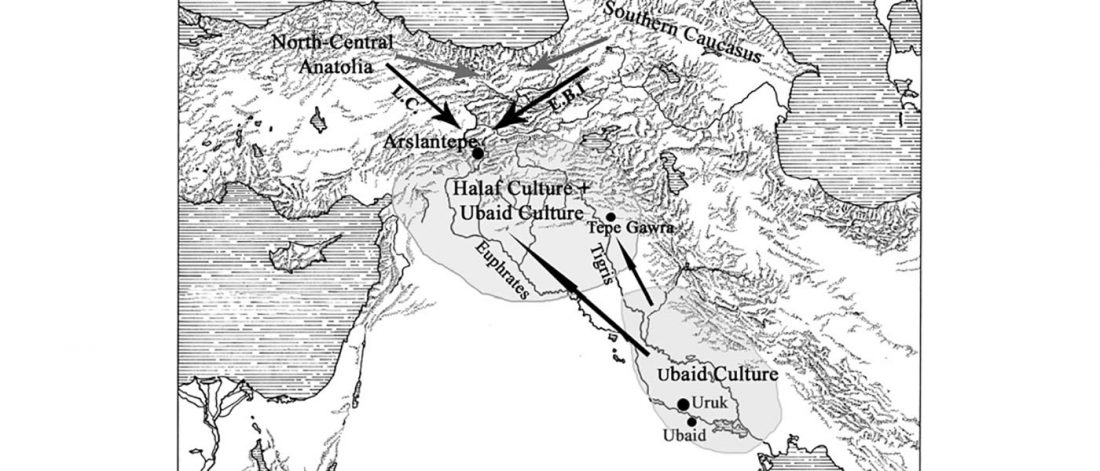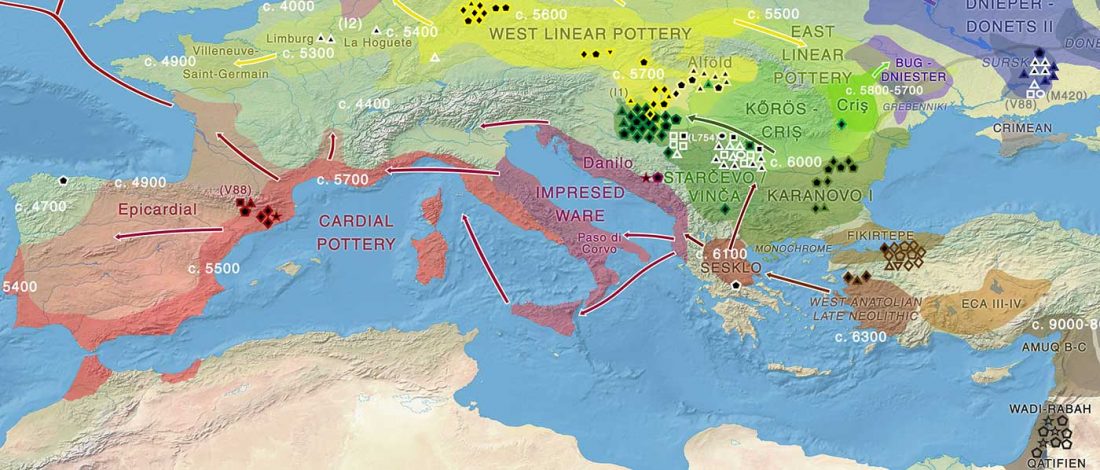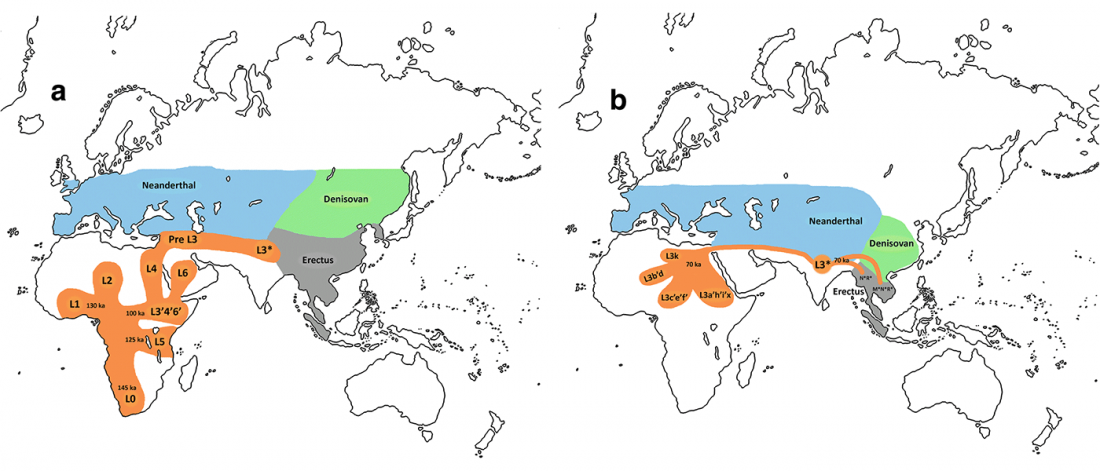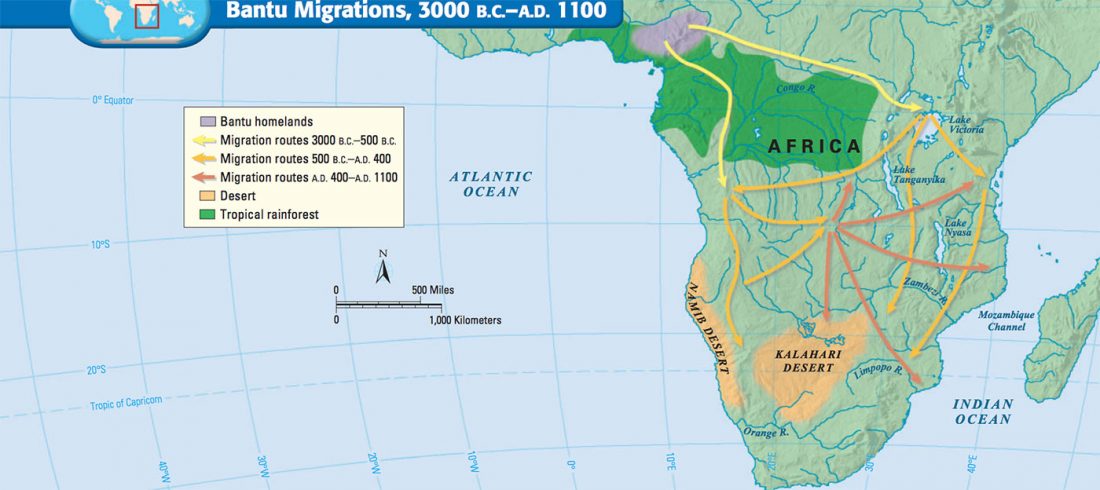
Open access African evolutionary history inferred from whole genome sequence data of 44 indigenous African populations, by Fan et al. Genome Biology (2019) 20:82.
Interesting excerpts (emphasis mine):
Introduction
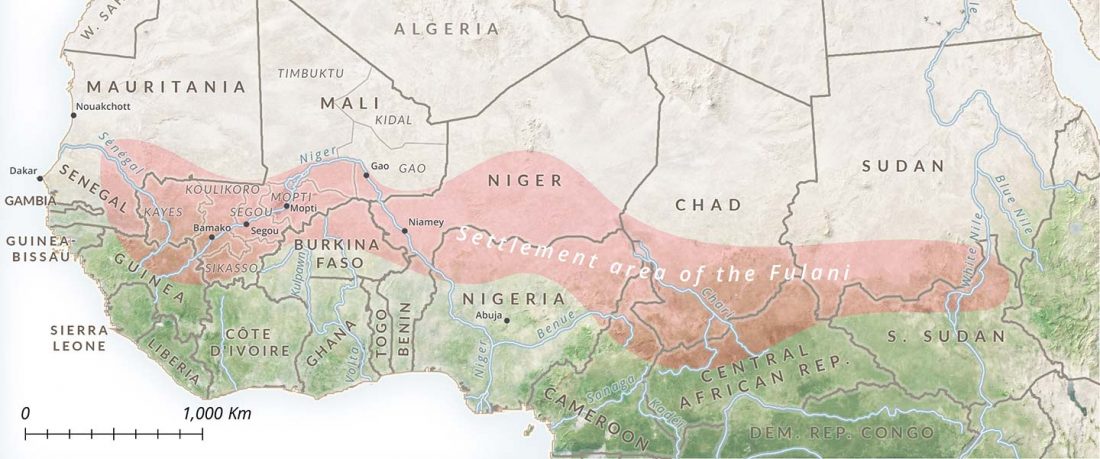
… Read the rest “Fulani from Cameroon show ancestry similar to Afroasiatic speakers from East Africa”To extend our knowledge of patterns of genomic diversity in Africa, we generated high coverage (30×) genome sequencing data from 43 geographically diverse Africans originating from 22 ethnic groups, representing a broad array of ethnic, linguistic, cultural, and geographic diversity (Additional file 1: Table S1). These include a number of populations of anthropological interest that have never previously been characterized for high-coverage genome sequence diversity such as Afroasiatic-speaking El