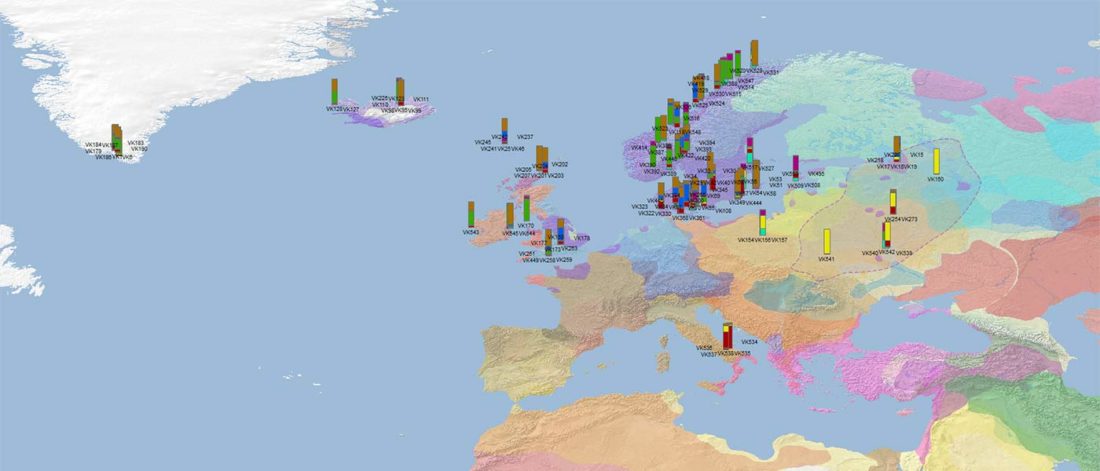
Recent paper (behind paywall) Population genomics of the Viking world, by Margaryan et al. Nature (2020), containing almost exactly the same information as its bioRxiv preprint.
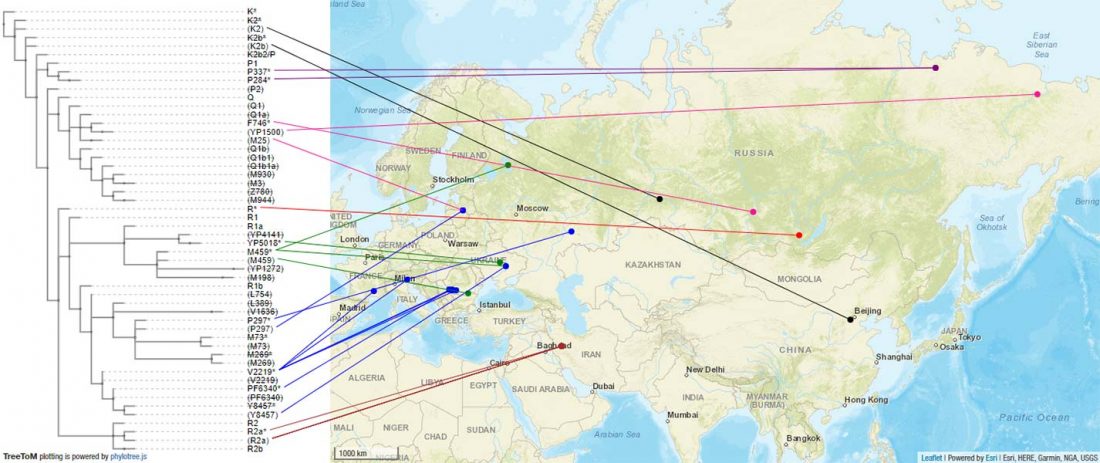
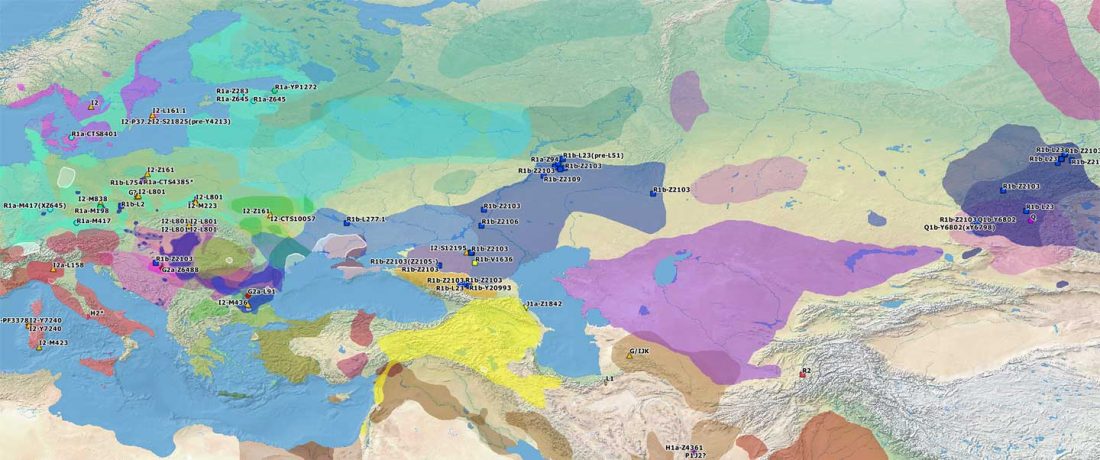
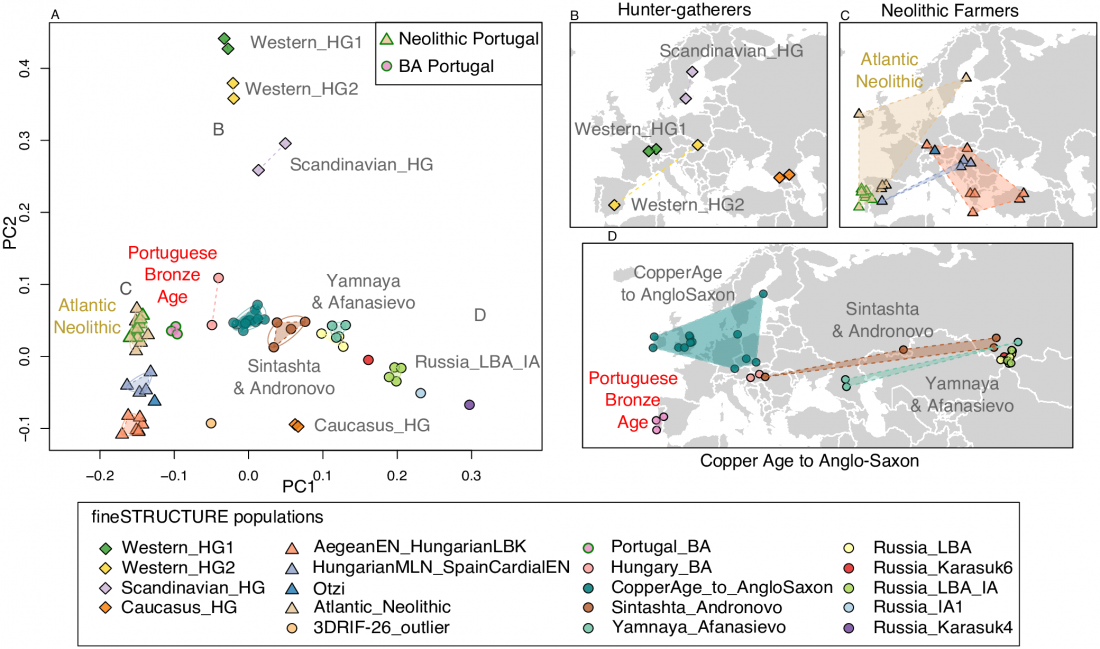
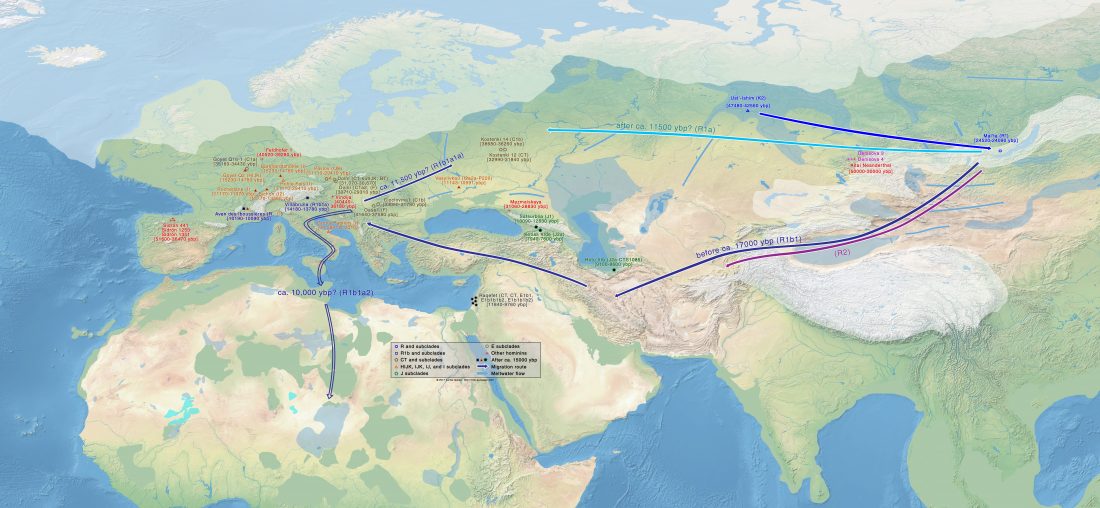
I have used Y-SNP inferences recently reported by FTDNA (see below) to update my Ancient DNA Dataset and the ArcGIS Online Map, and also to examine the chronological and geographical evolution of Y-DNA (alone and in combination with ancestry).
Sections of this post:
- Iron Age to Medieval Y-DNA
- Iron Age to Medieval Ancestry
- Iron Age to Medieval Y-DNA + Ancestry
- FTDNA’s big public debut