The following are some recent developments and updates:
I. Ancient DNA Dataset version 2
I.1. Accurate mtDNA haplogroups
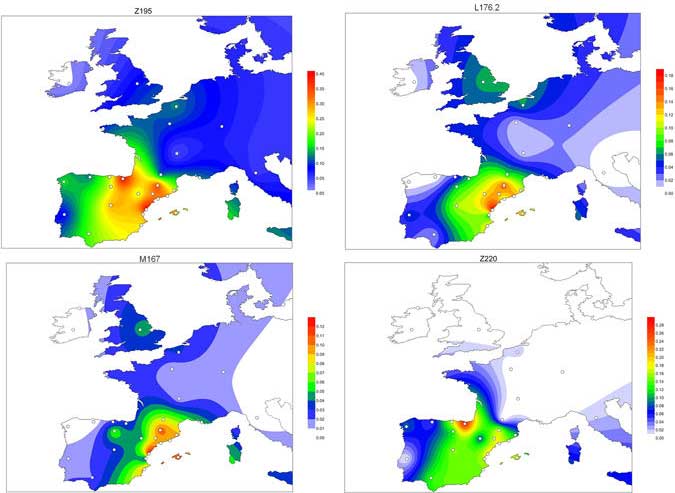
I was meaning to update the mtDNA part of the Ancient DNA Dataset, and finally found some time to review FTDNA and YFull nomenclature (including hyperlinks), as well as those SNP calls from published samples found in YFull’s MTree. So, if you are interested in studies of mtDNA phylogeography, I think the data is now accurate and much more useful.
Given the number of columns and the size of the files, I have decided to post shorter standard versions, by … Read the rest “mtDNA, lactase persistence, and admixr for ADMIXTOOLS”