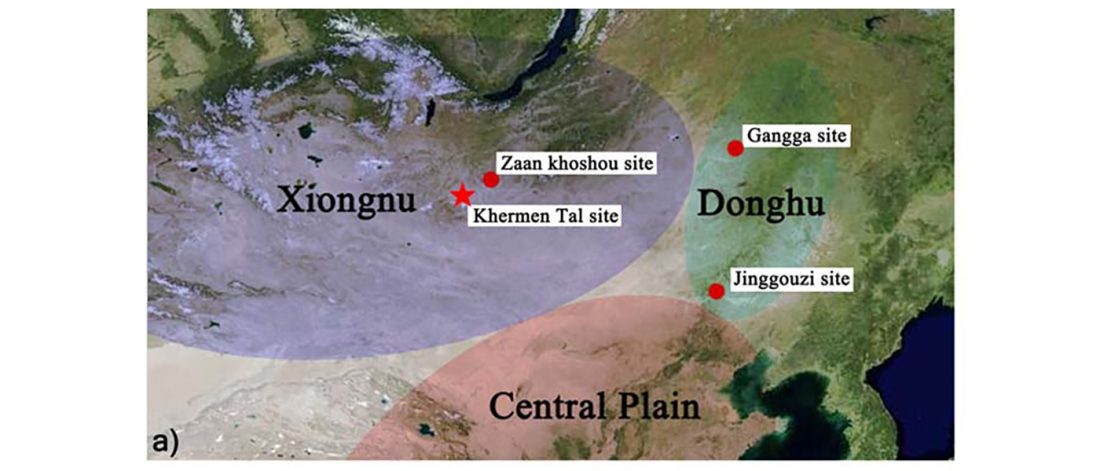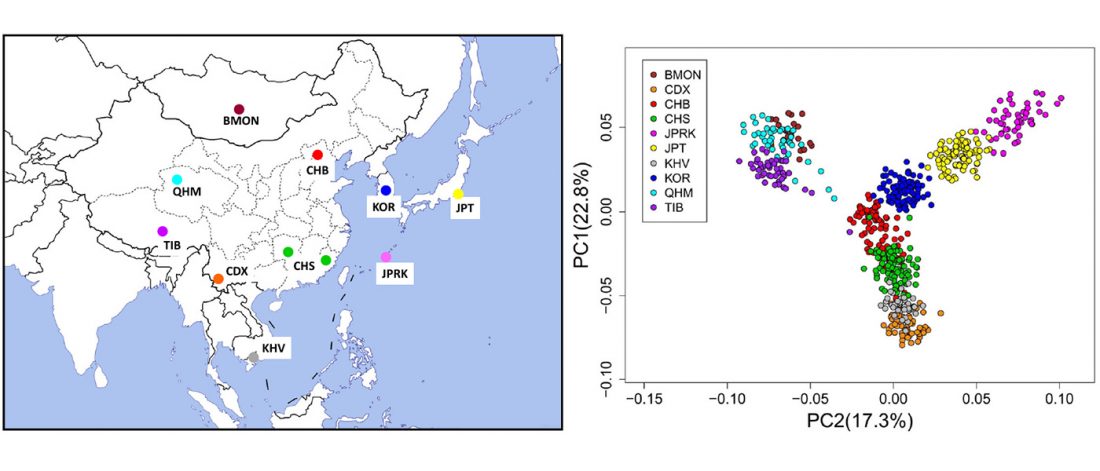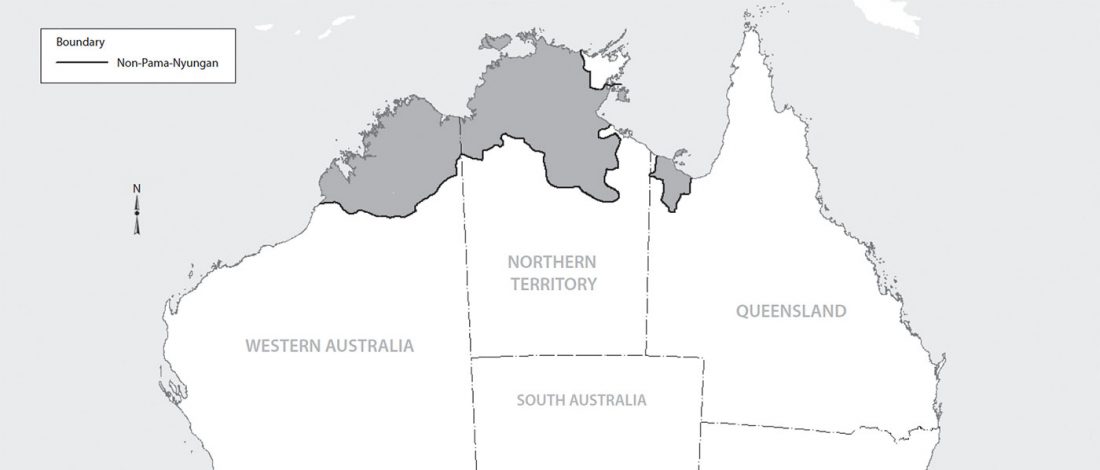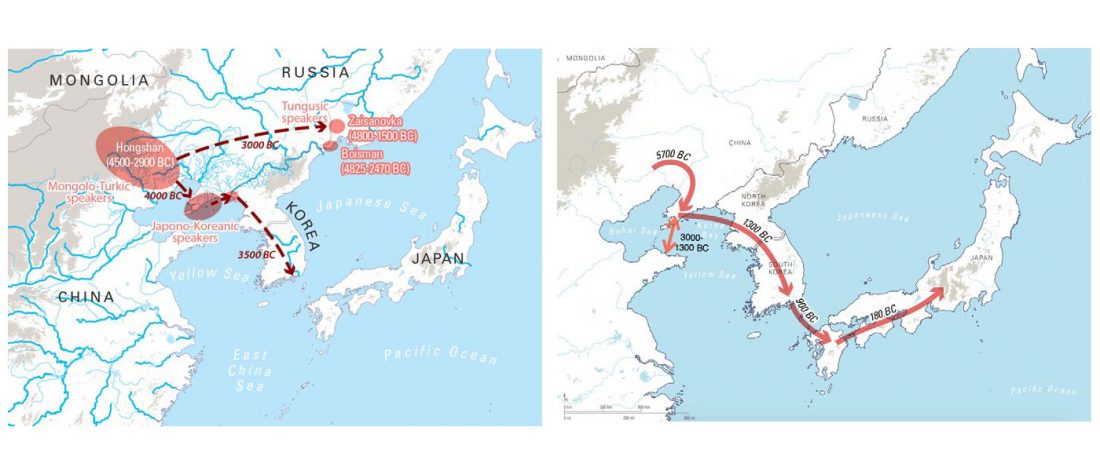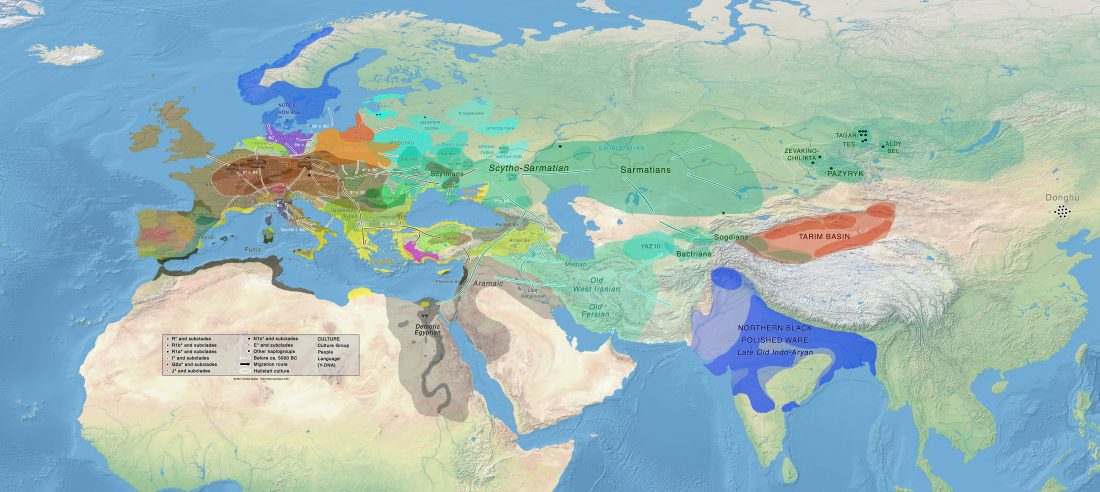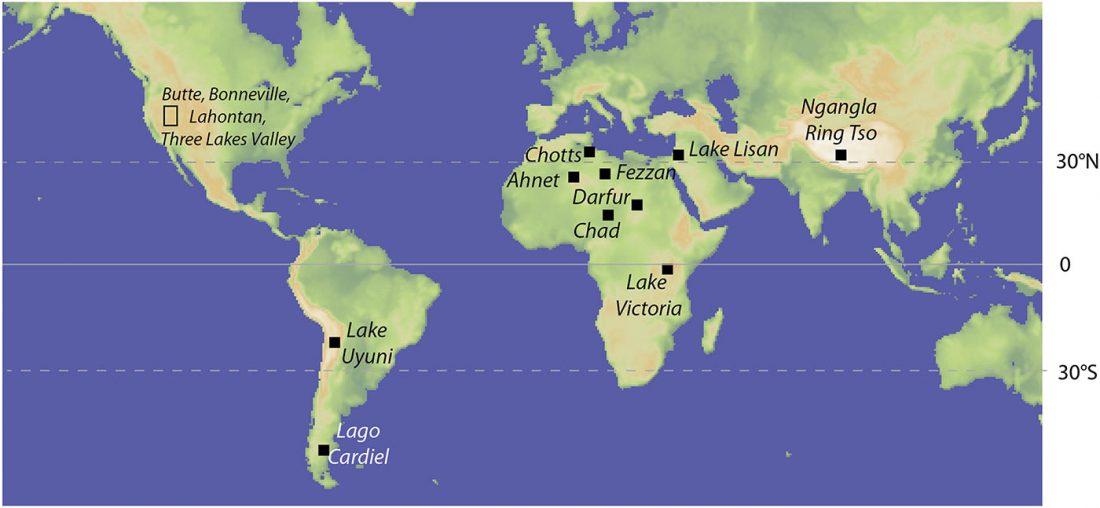
Interesting new paper (behind paywall) Megalakes in the Sahara? A Review, by Quade et al. (2018).
Abstract (emphasis mine):
… Read the rest “Sahara’s rather pale-green and discontinuous Sahelo-Sudanian steppe corridor, and the R1b – Afroasiatic connection”The Sahara was wetter and greener during multiple interglacial periods of the Quaternary, when some have suggested it featured very large (mega) lakes, ranging in surface area from 30,000 to 350,000 km2. In this paper, we review the physical and biological evidence for these large lakes, especially during the African Humid Period (AHP) 11–5 ka. Megalake systems from around the world provide a checklist of diagnostic features, such as multiple well-defined shoreline benches, wave-rounded beach gravels where coarse material is