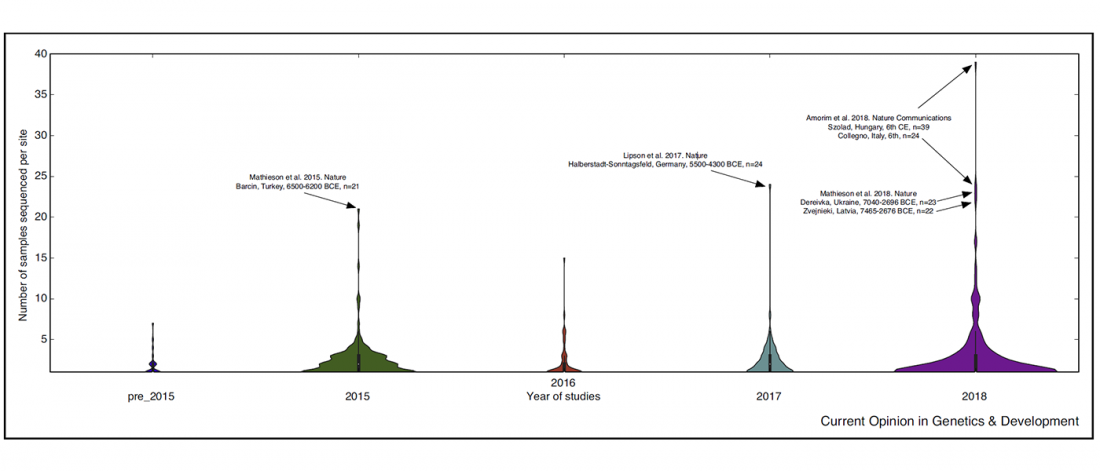
Short review (behind paywall) The importance of fine-scale studies for integrating paleogenomics and archaeology, by Krishna R. Veeramah, Current Opinion in Genetics & Development (2018) 53:83-89.
Abstract (emphasis mine):
… Read the rest “The importance of fine-scale studies for integrating palaeogenomics and archaeology”There has been an undercurrent of intellectual tension between geneticists studying human population history and archaeologists for almost 40 years. The rapid development of paleogenomics, with geneticists working on the very material discovered by archaeologists, appears to have recently heightened this tension. The relationship between these two fields thus far has largely been of a multidisciplinary nature, with archaeologists providing the raw materials for sequencing, as well as a scaffold