New paper Biparental Inheritance of Mitochondrial DNA in Humans, by Luo et al. PNAS (2018).
Interesting excerpts (emphasis mine):
Abstract
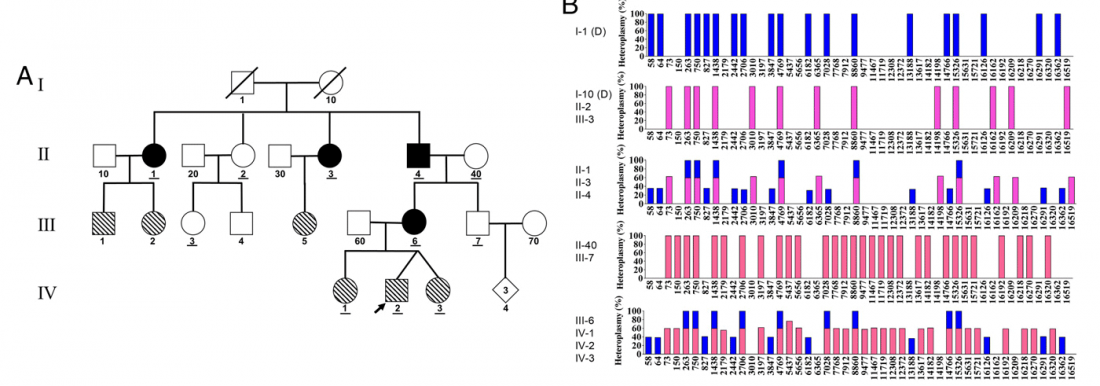
… Read the rest “Biparental inheritance of mitochondrial DNA in humans”Although there has been considerable debate about whether paternal mitochondrial DNA (mtDNA) transmission may coexist with maternal transmission of mtDNA, it is generally believed that mitochondria and mtDNA are exclusively maternally inherited in humans. Here, we identified three unrelated multigeneration families with a high level of mtDNA heteroplasmy (ranging from 24 to 76%) in a total of 17 individuals. Heteroplasmy of mtDNA was independently examined by high-depth whole mtDNA sequencing analysis in our research laboratory and in two Clinical