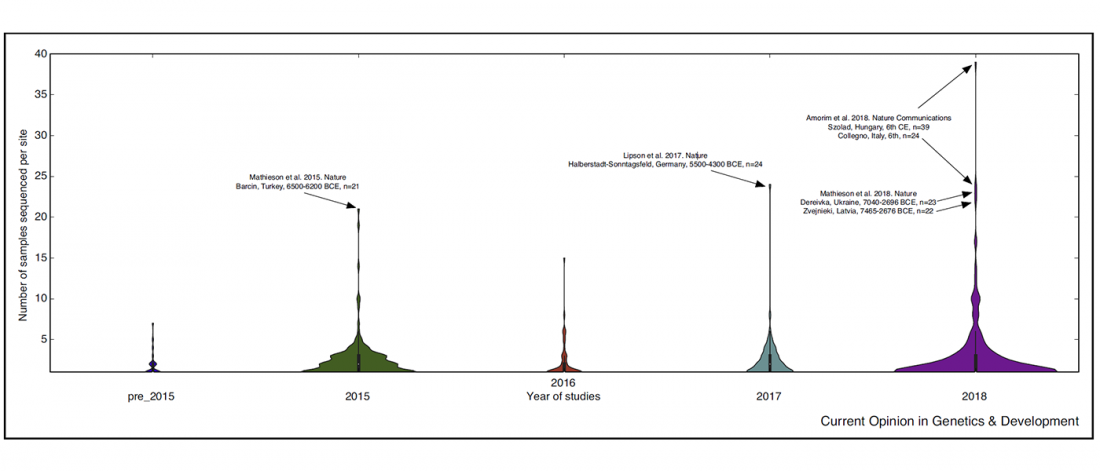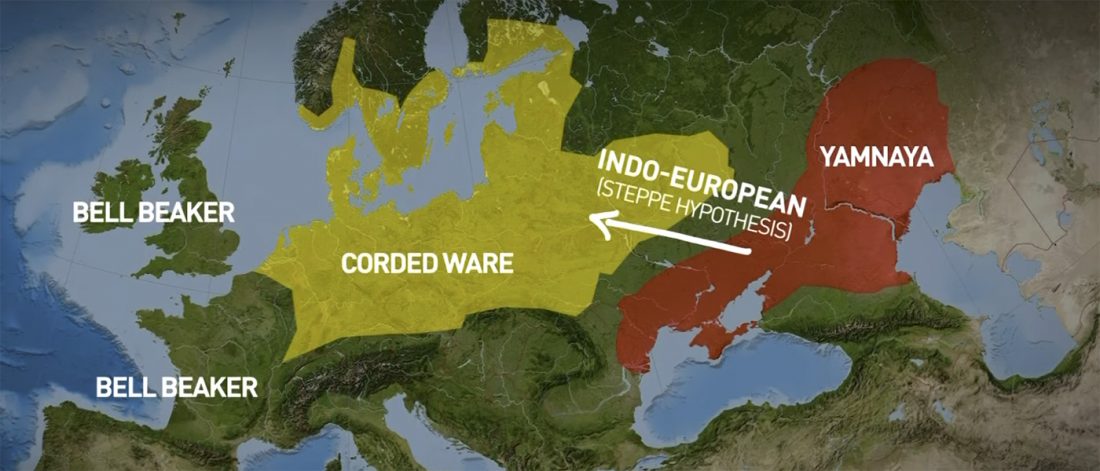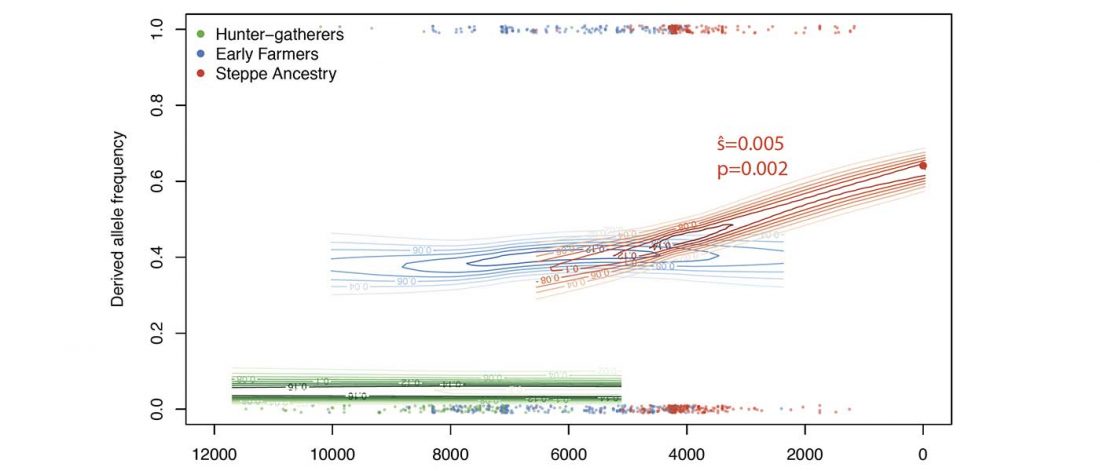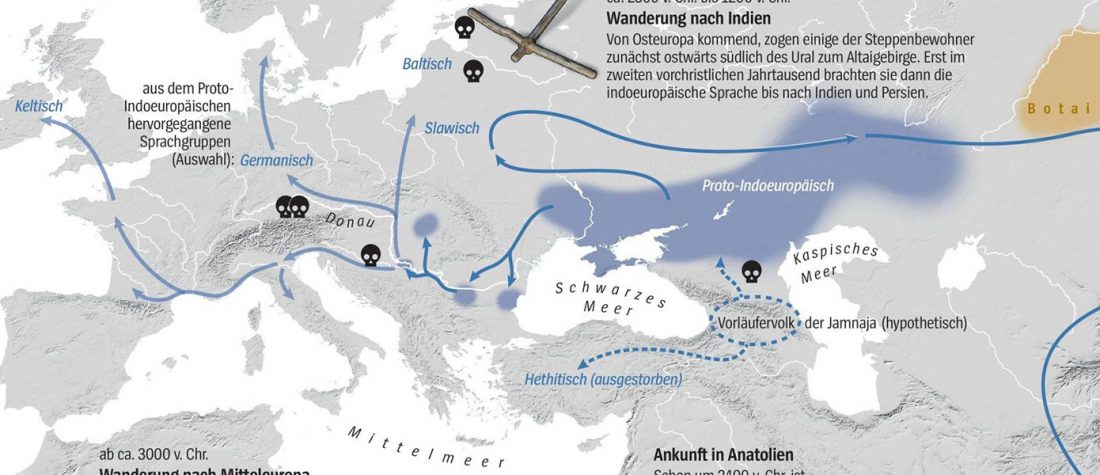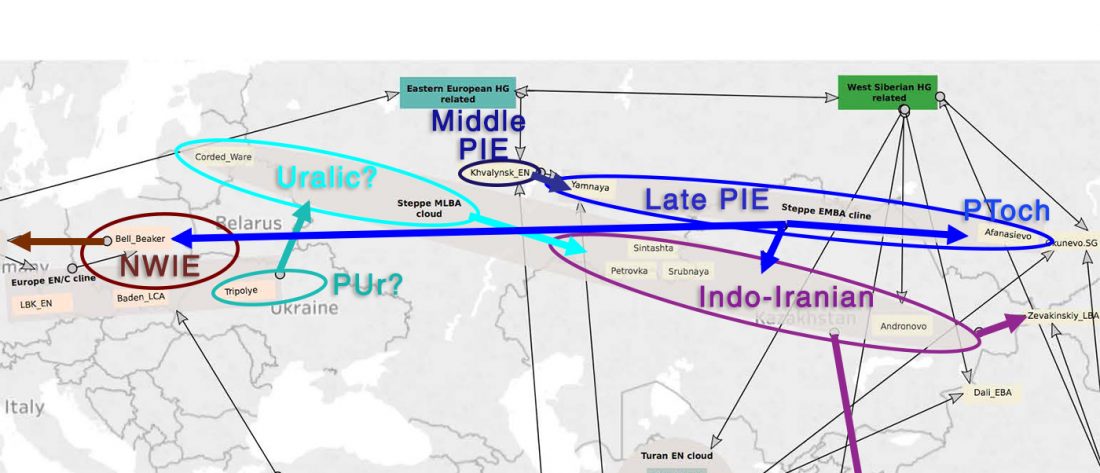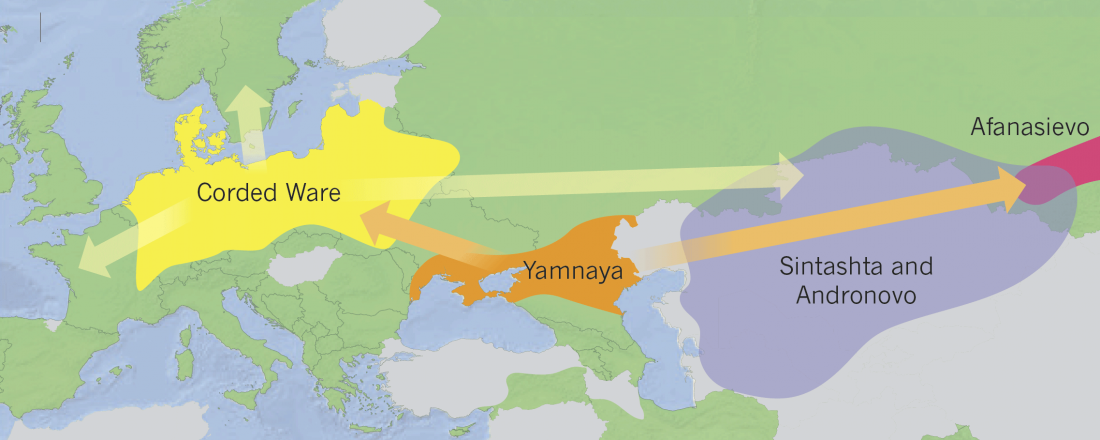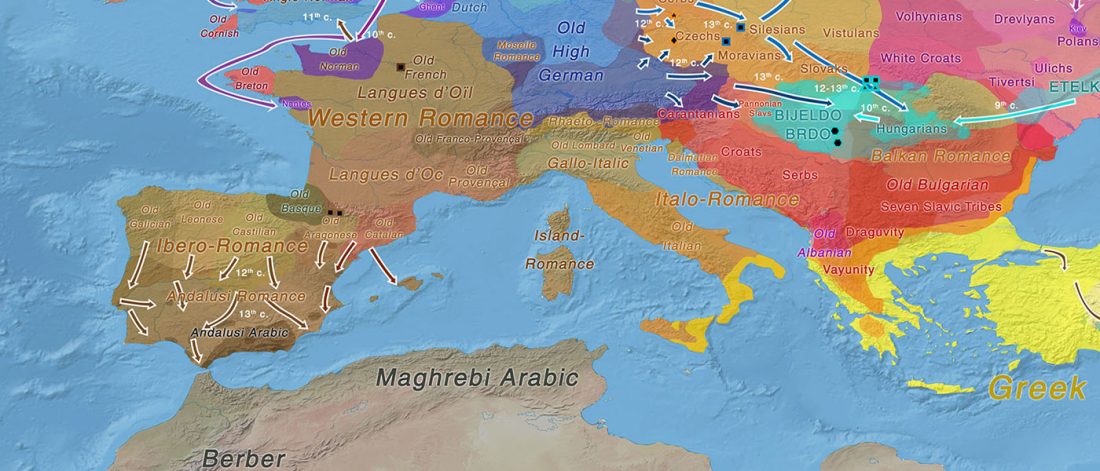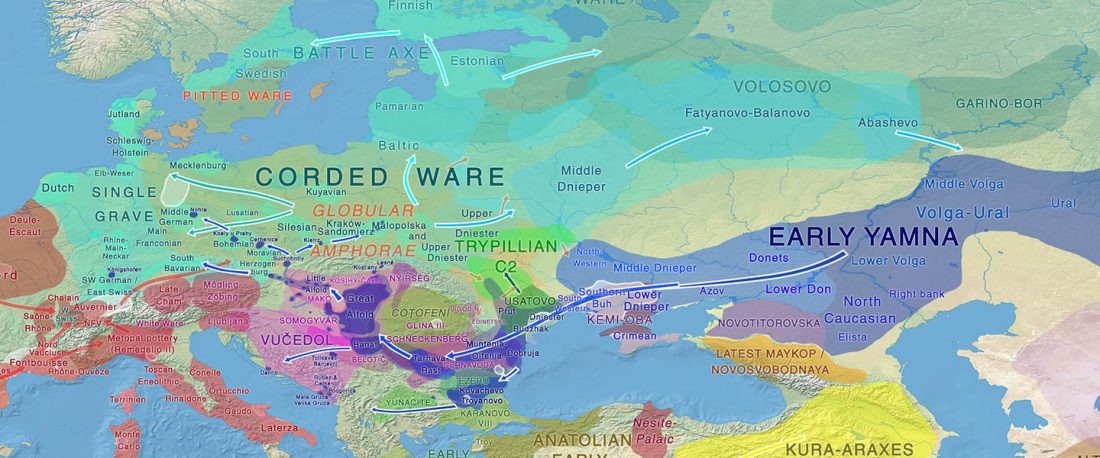
There is a good reason for hope, for those who look for a happy ending to the revolution of population genomics that is quickly turning into an involution led by beliefs and personal interests. This blog is apparently one of the the most read sites on Indo-European peoples, if not the most read one, and now on Uralic peoples, too.
I’ve been checking the analytics of our sites, and judging by the numbers of the English blog, Indo-European.eu (without the other languages) is quickly turning into the most visited one from Academia Prisca‘s sites on Indo-European languages, beyond … Read the rest “Ahead of the (Indo-European – Uralic) game: in theory and in numbers”