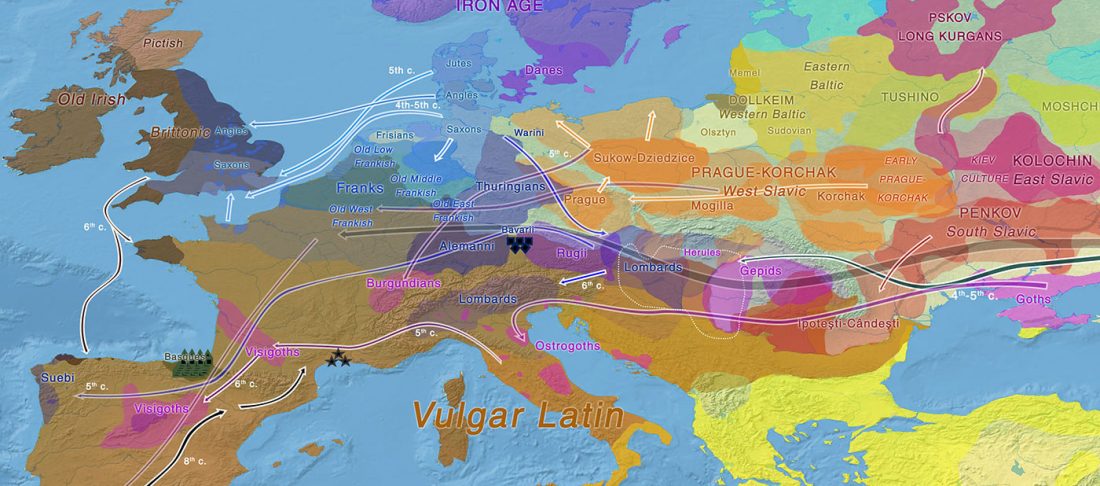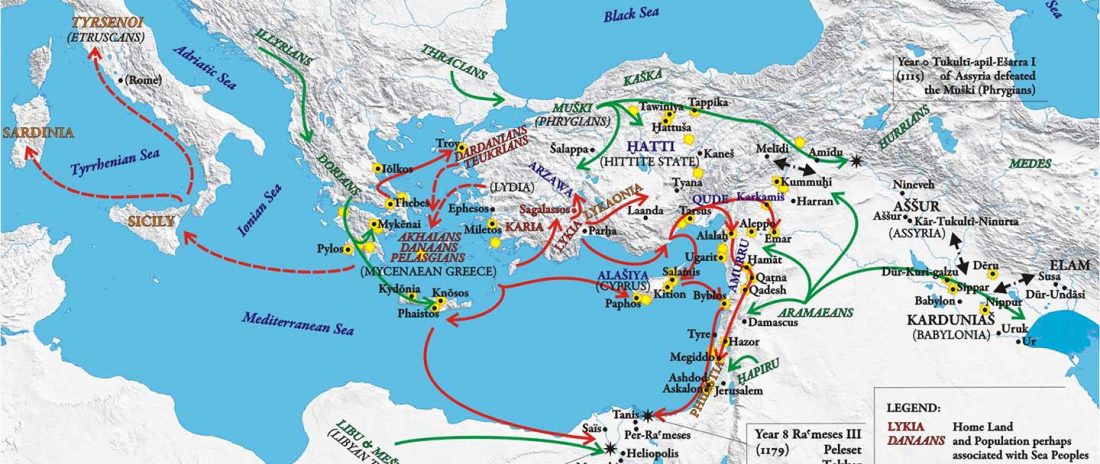
New open access paper Ancient DNA sheds light on the genetic origins of early Iron Age Philistines, by Feldman et al. Science Advances (2019) 5(7):eaax0061.
Interesting excerpts (modified for clarity, emphasis mine):
… Read the rest “Sea Peoples behind Philistines were Aegeans, including R1b-M269 lineages”Here, we report genome-wide data from human remains excavated at the ancient seaport of Ashkelon, forming a genetic time series encompassing the Bronze to Iron Age transition. We find that all three Ashkelon populations derive most of their ancestry from the local Levantine gene pool. The early Iron Age population was distinct in its high genetic affinity to European-derived populations and in the high variation of that