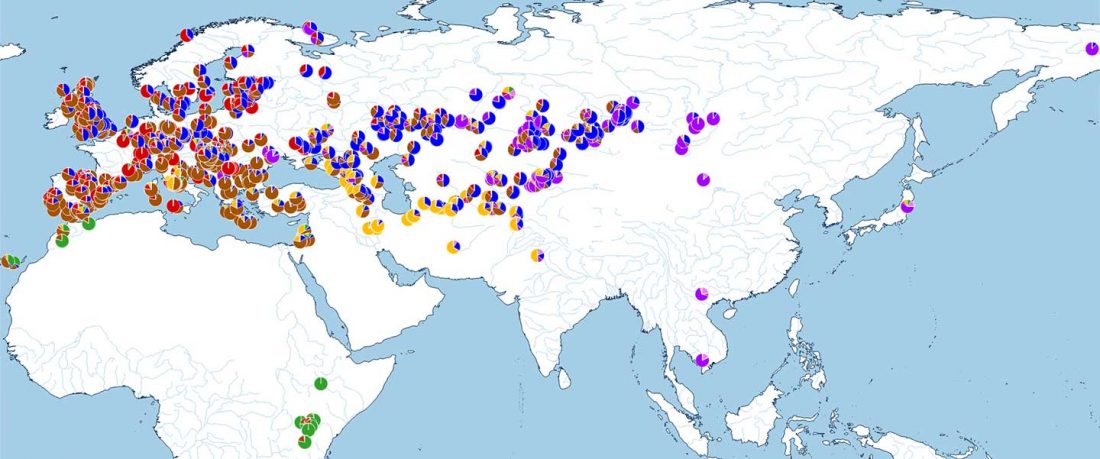
The following are updated files for unsupervised ADMIXTURE of most available ancient Eurasian samples with K=7. For reference, see PCA of ancient and modern Eurasian samples.
NOTE. For a precise interpretation of ancestry evolution, be sure to first check the posts on the expansion of “Steppe ancestry”, on the spread of Yamnaya ancestry with Indo-Europeans, and on the evolution of Corded Ware ancestry typical of modern Uralic populations.
ADMIXTURE timeline
This is a YouTube video similar to the one on Indo-Europeans and Y-DNA evolution:
Some comments
- I have tried running supervised ADMIXTURE models by selecting