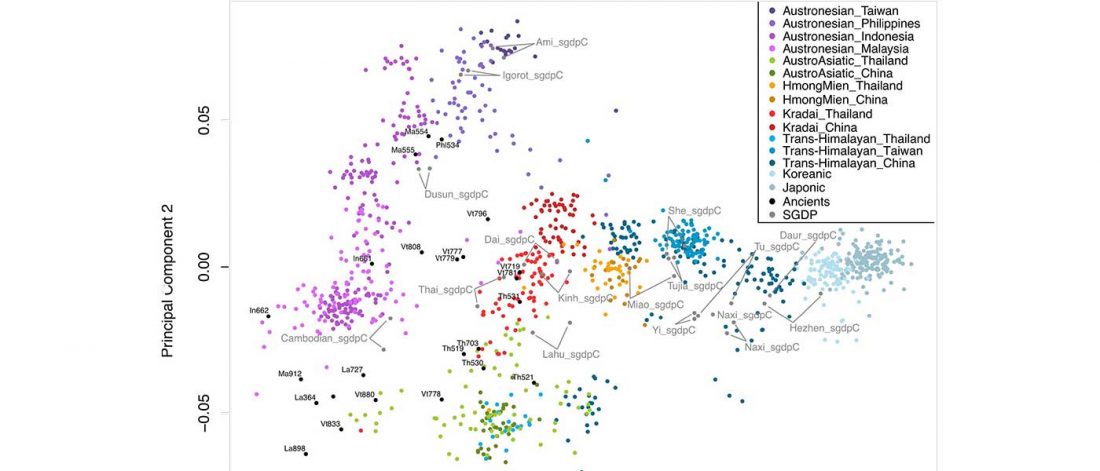
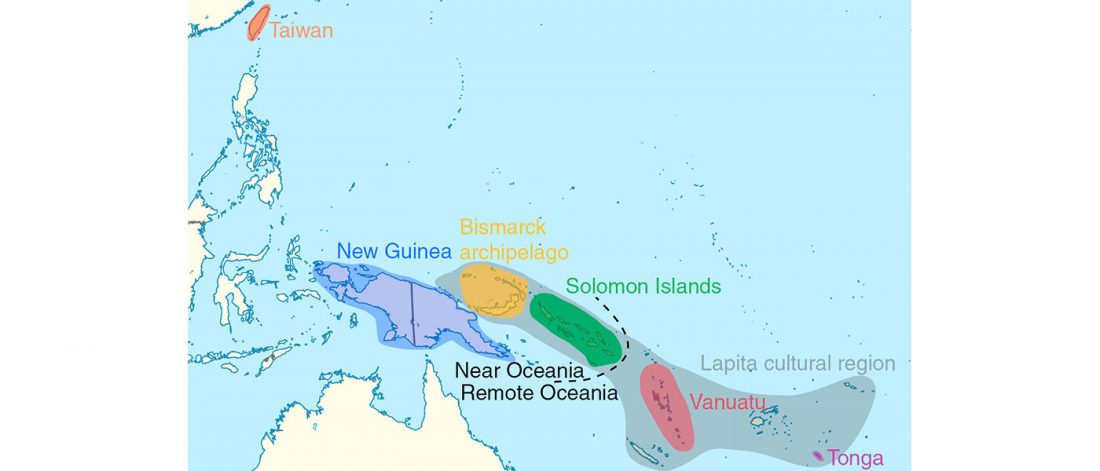
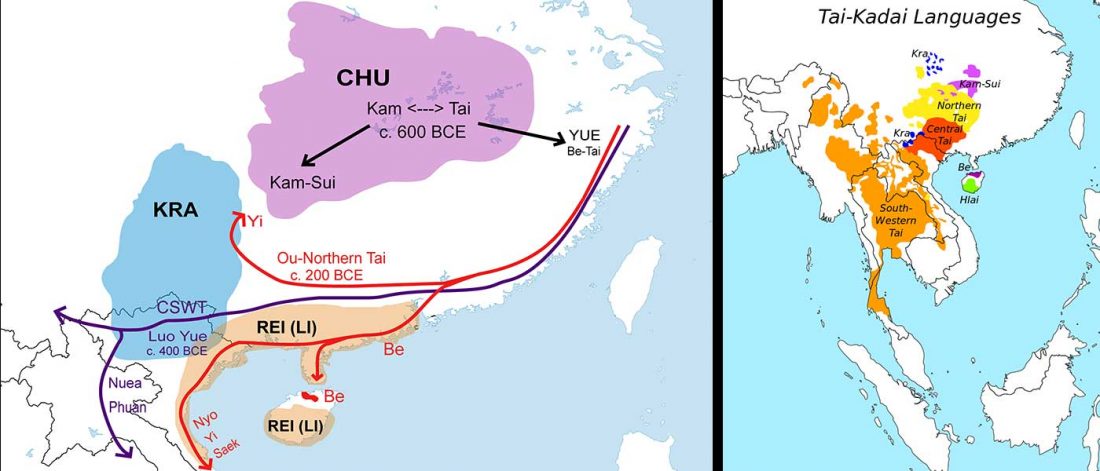
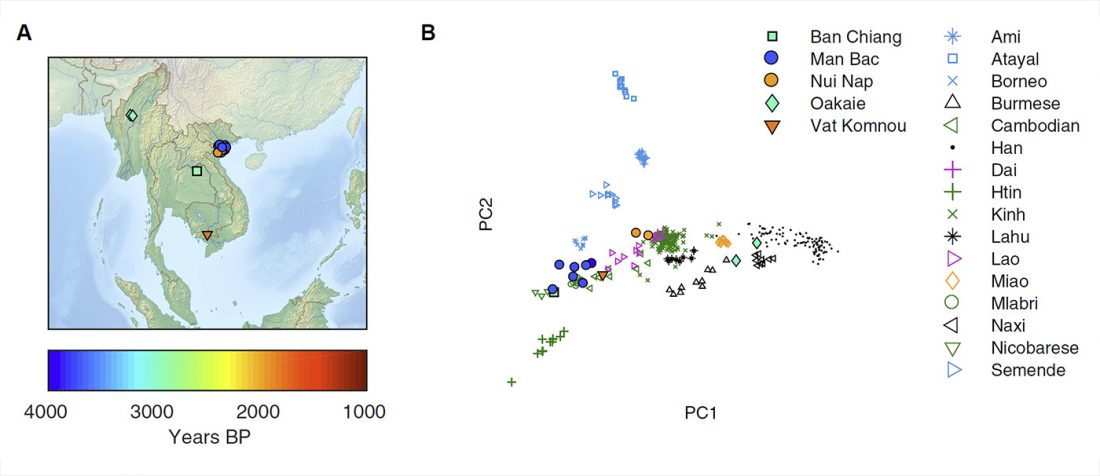
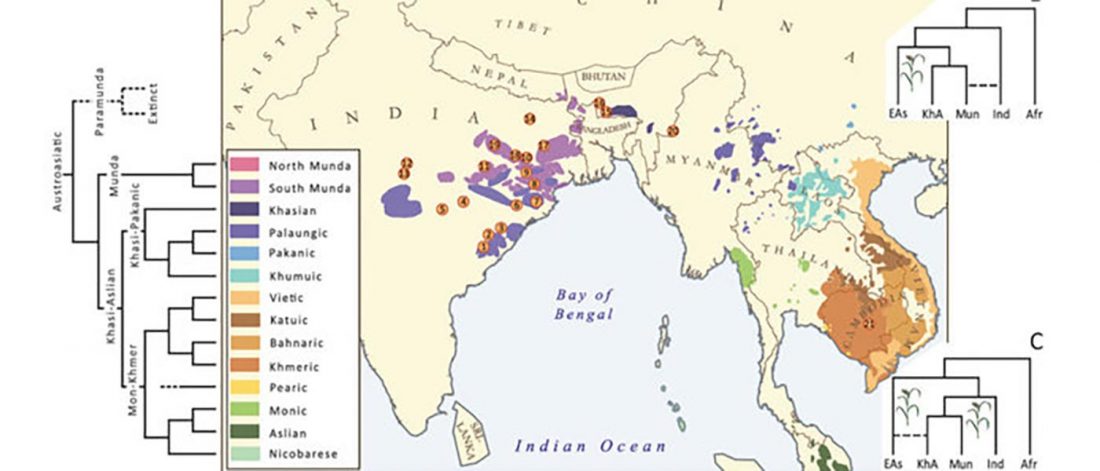
Munda admixture happened probably during the ANI-ASI mixture
Preprint The genetic legacy of continental scale admixture in Indian Austroasiatic speakers, by Tätte et al. bioRxiv (2018).
Interesting excerpts:
… Read the rest “Munda admixture happened probably during the ANI-ASI mixture”Studies analysing mtDNA and Y chromosome markers have revealed a sex-specific admixture pattern of admixture of Southeast and South Asian ancestry components for Munda speakers. While close to 100% of mtDNA lineages present in Mundas match those in other Indian populations, around 65% of their paternal genetic heritage is more closely related to Southeast Asian than South Asian variation. Such a contrasting distribution of maternal and paternal lineages among the Munda speakers is a classic example of ‘father tongue