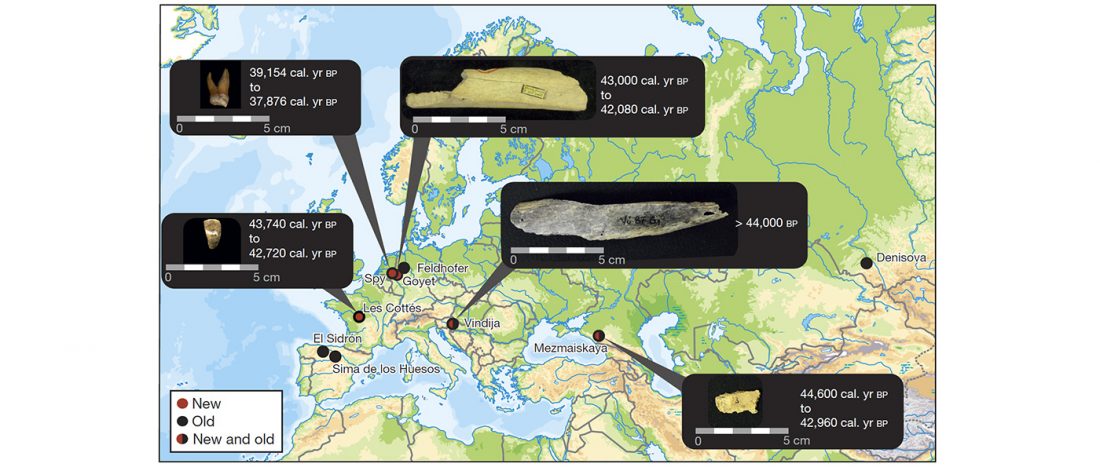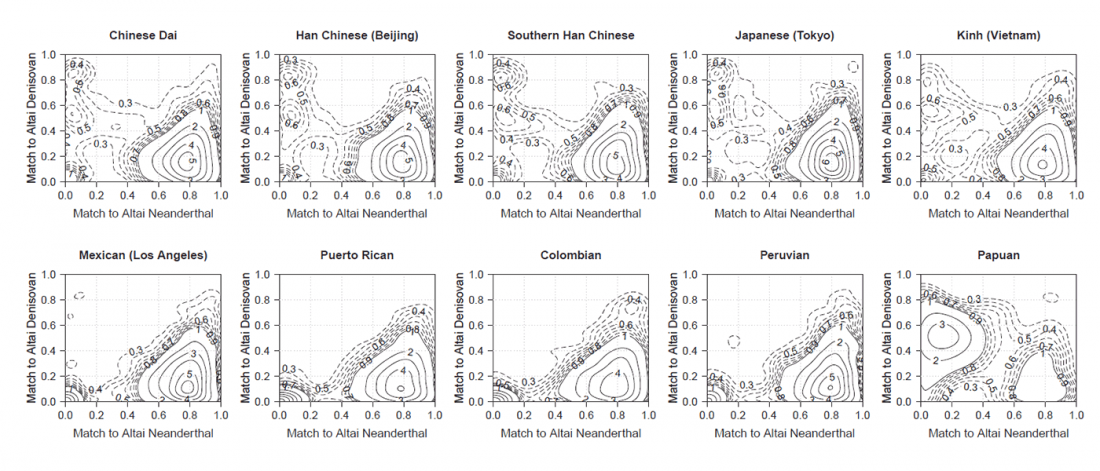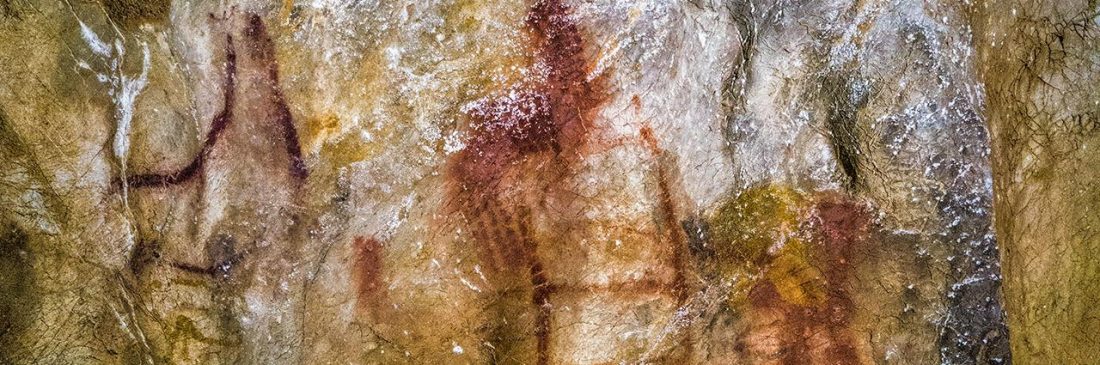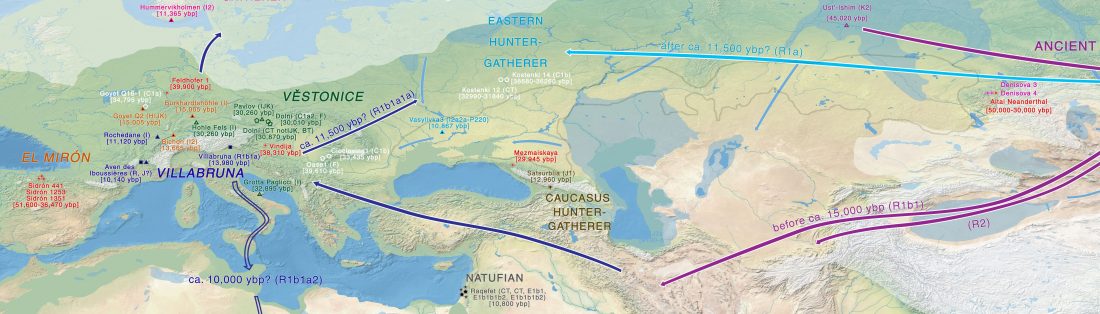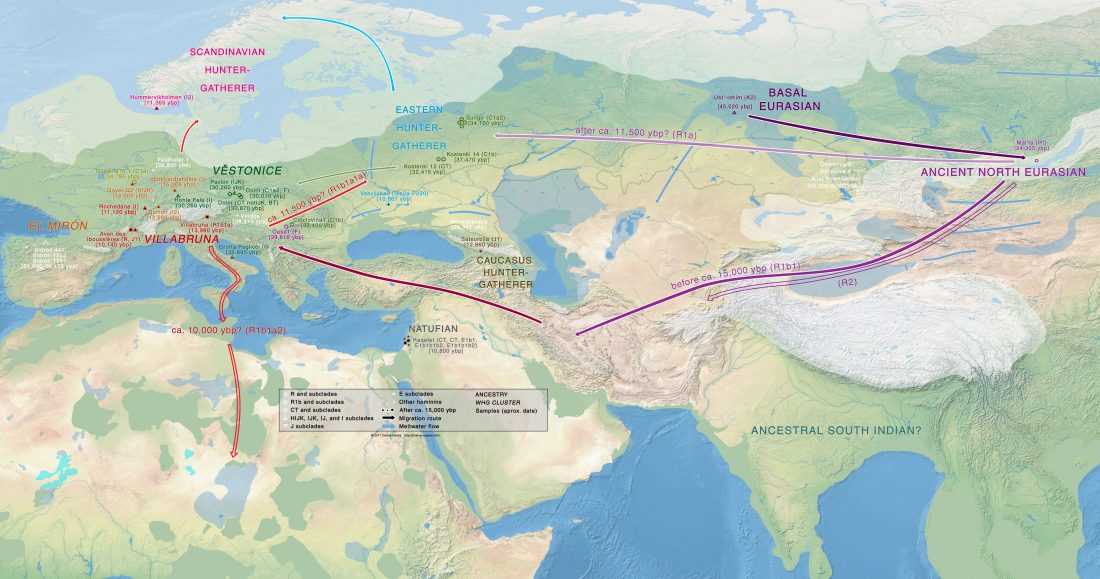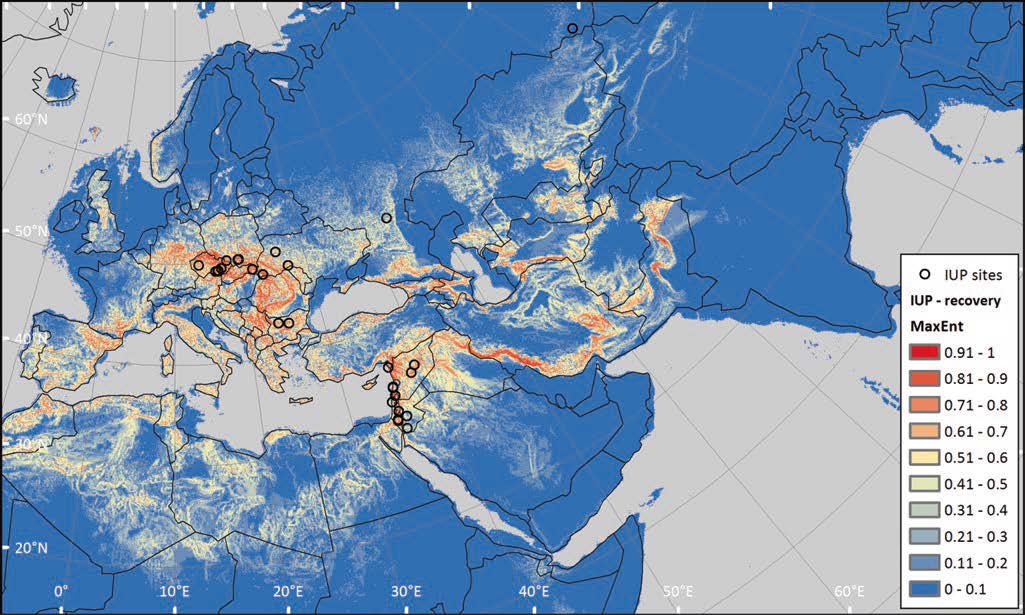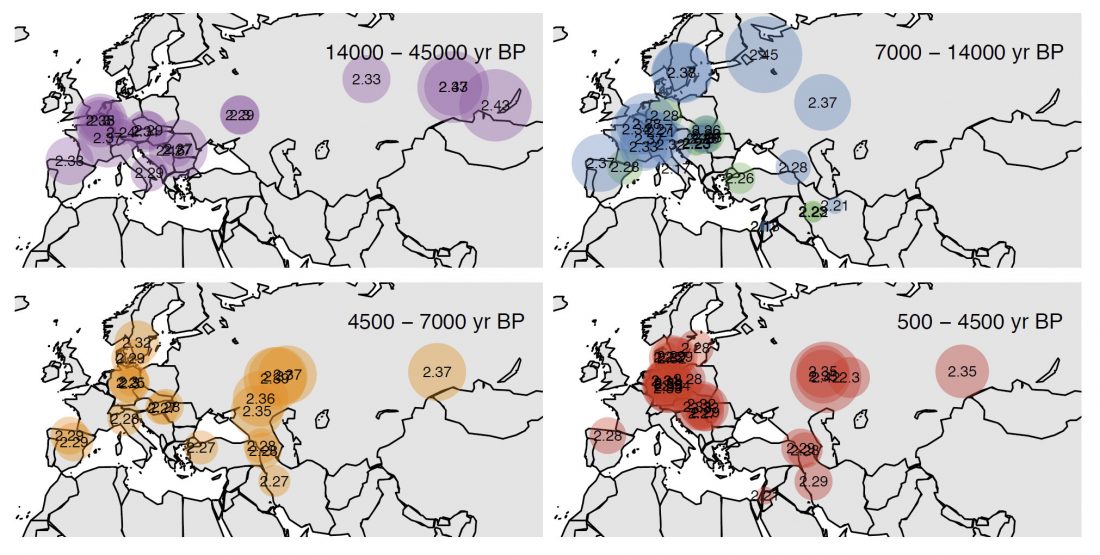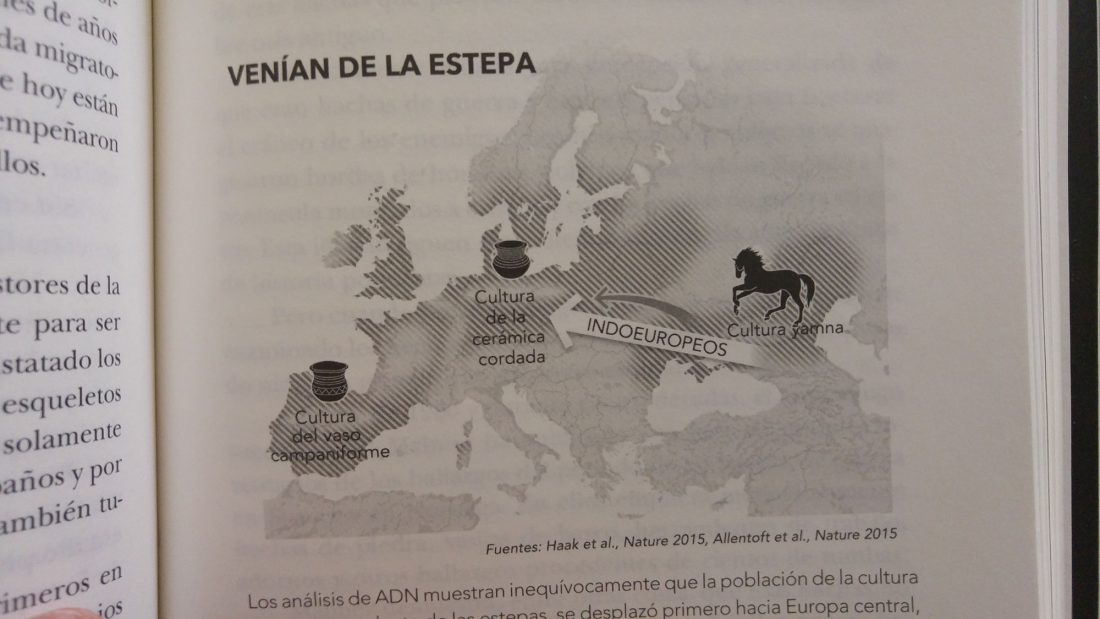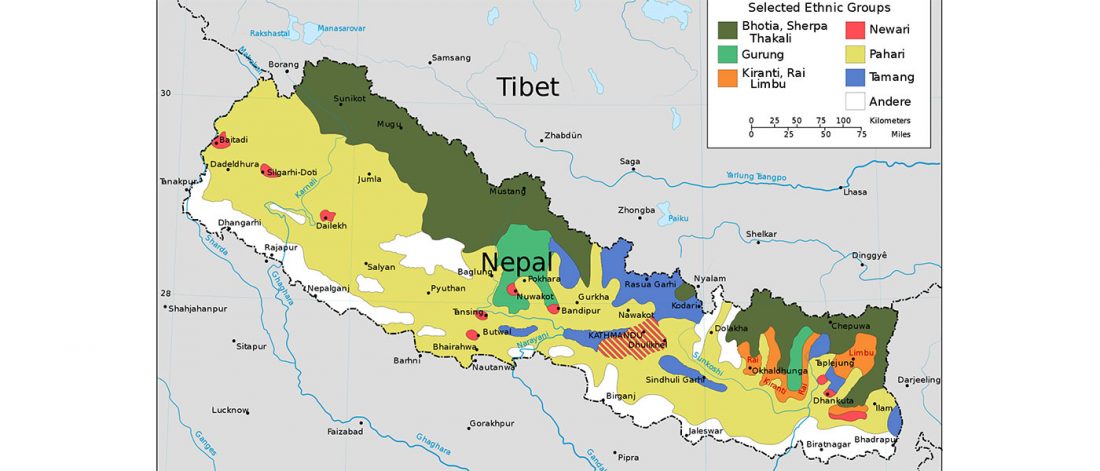
Open access Demographic history and genetic adaptation in the Himalayan region inferred from genome-wide SNP genotypes of 49 populations, by Arciero et al. Mol. Biol. Evol (2018), accepted manuscript (msy094).
Abstract (emphasis mine):
… Read the rest “Demographic history and genetic adaptation in the Himalayan region”We genotyped 738 individuals belonging to 49 populations from Nepal, Bhutan, North India or Tibet at over 500,000 SNPs, and analysed the genotypes in the context of available worldwide population data in order to investigate the demographic history of the region and the genetic adaptations to the harsh environment. The Himalayan populations resembled other South and East Asians, but in addition displayed their own specific