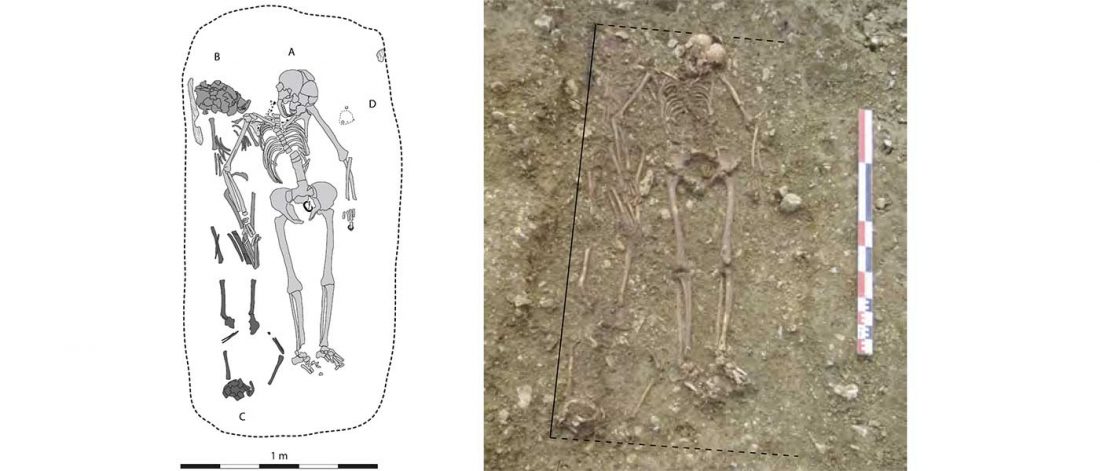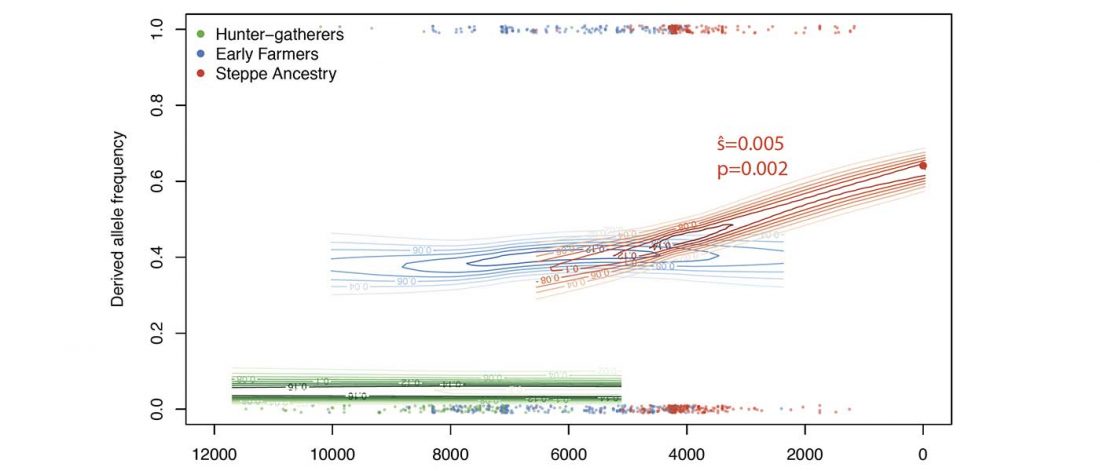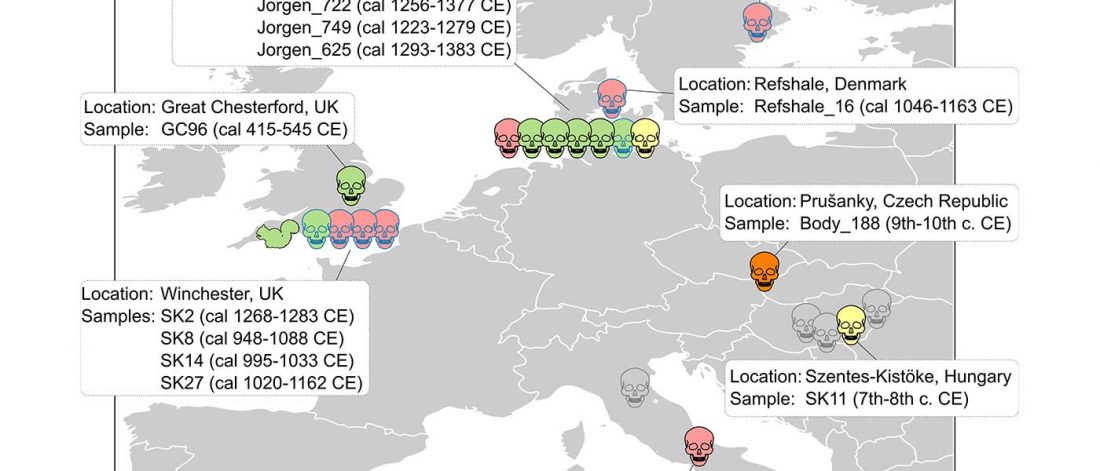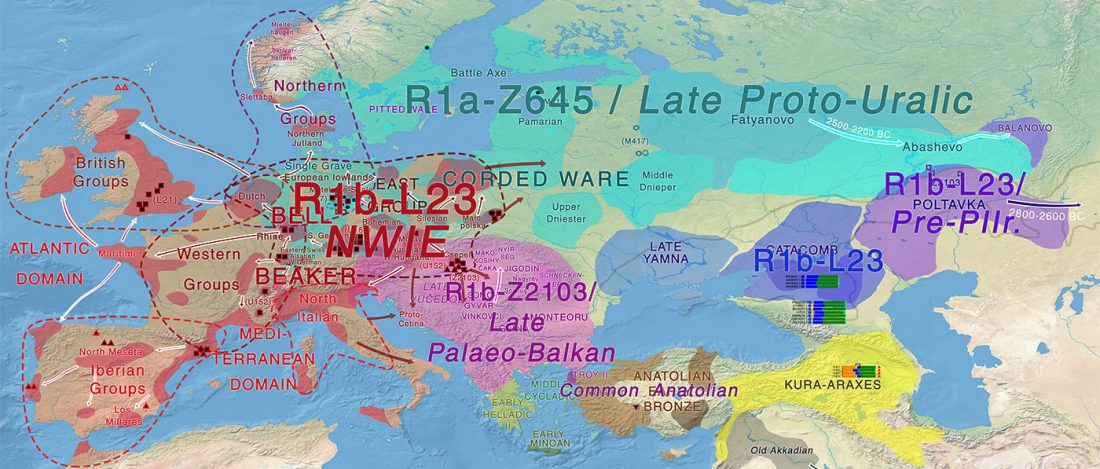
New paper (behind paywall) Estimating the age of p.(Phe508del) with family studies of geographically distinct European populations and the early spread of cystic fibrosis, by Farrell et al., European Journal of Human Genetics (2018).
Interesting excerpts (emphasis mine):
… Read the rest “Cystic fibrosis probably spread with expanding Bell Beakers”Our results revealed tMRCA average values ranging from 4725 to 1175 years ago and support the estimates of Serre et al. (3000–6000 years ago) [11], rather than Morral et al. (52,000 years ago) [6], but the latter figure was challenged by Kaplan et al. [26] because of disagreement with assumptions used in their calculations. In addition, the tMRCA values from western