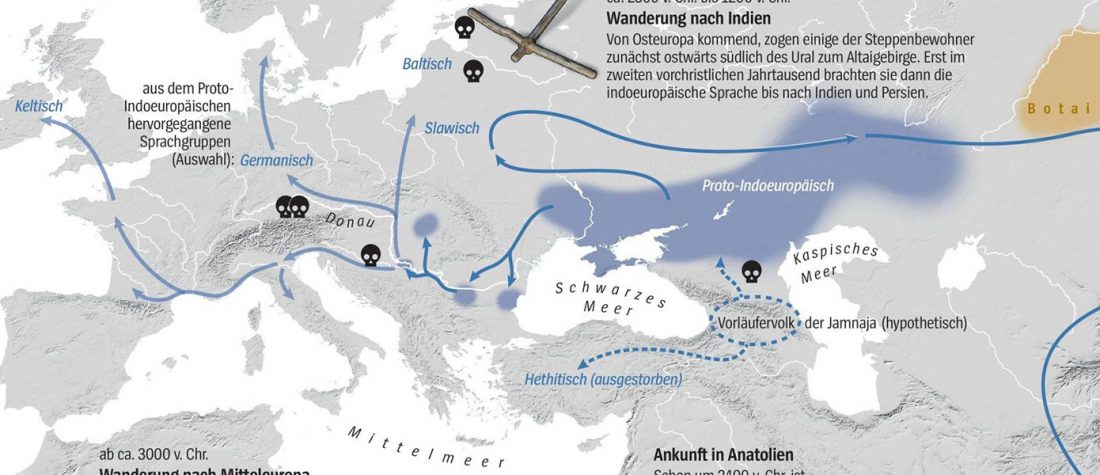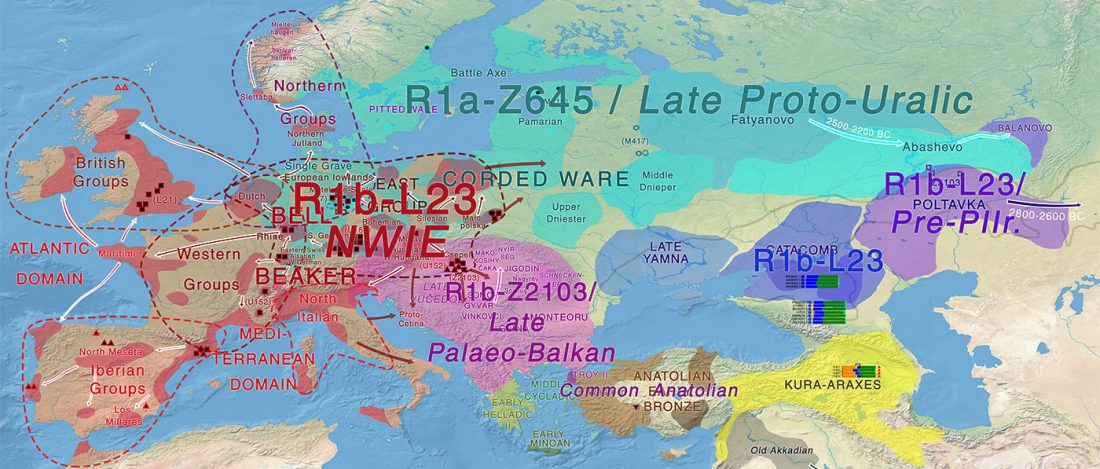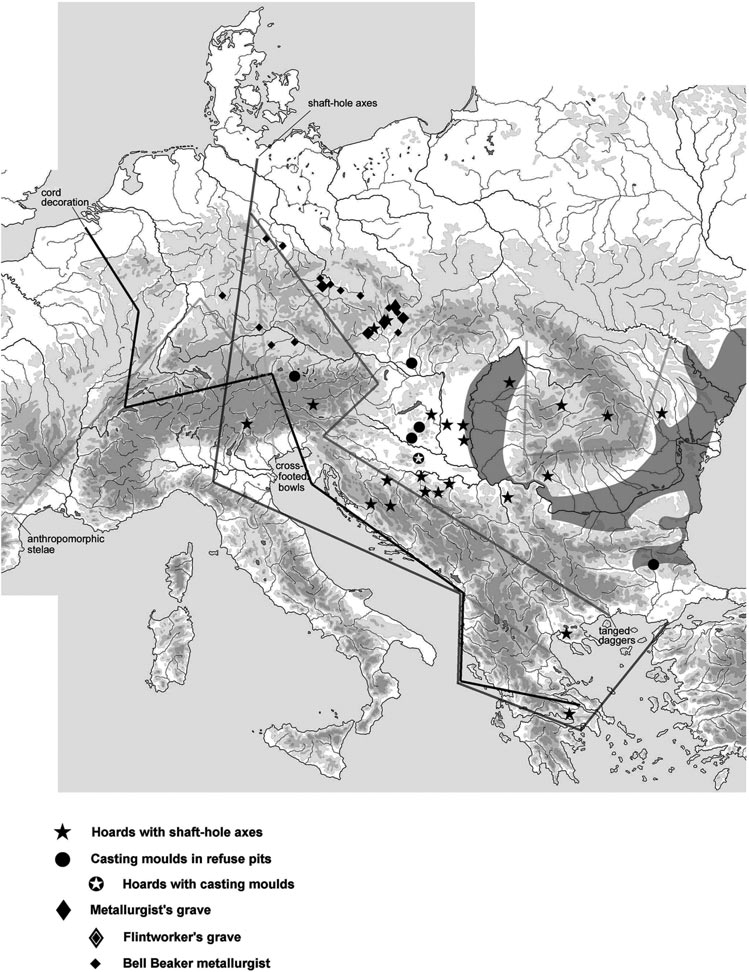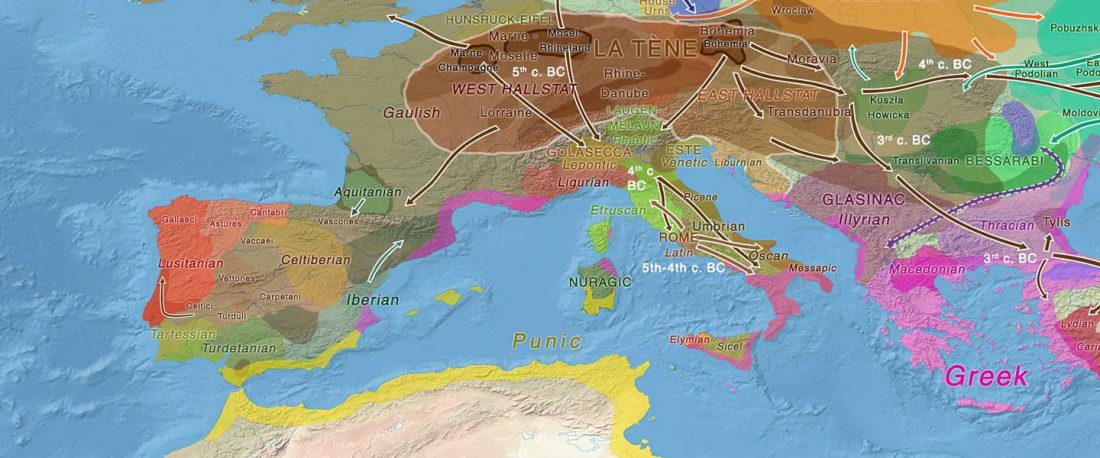
The recent data on ancient DNA from Iberia published by Olalde et al. (2019) was interesting for many different reasons, but I still have the impression that the authors – and consequently many readers – focused on not-so-relevant information about more recent population movements, or even highlighted the least interesting details related to historical events.
I have already written about the relevance of its findings for the Indo-European question in an initial assessment, then in a more detailed post about its consequences, then about the arrival of Celtic languages with hg. R1b-M167, and later in combination with … Read the rest “North-West Indo-Europeans of Iberian Beaker descent and haplogroup R1b-P312”