Open access Genetic structure, divergence and admixture of Han Chinese, Japanese and Korean populations, by Wang, Lu, Chung, and Xu, Hereditas (2018) 155:19.
Abstract (emphasis mine):
… Read the rest “Genetic structure, divergence and admixture of Han Chinese, Japanese and Korean populations”Background
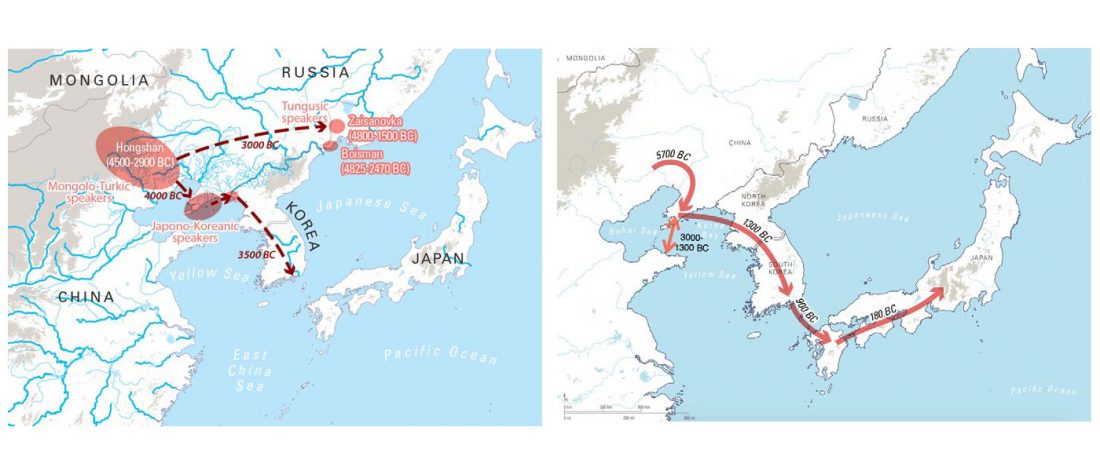
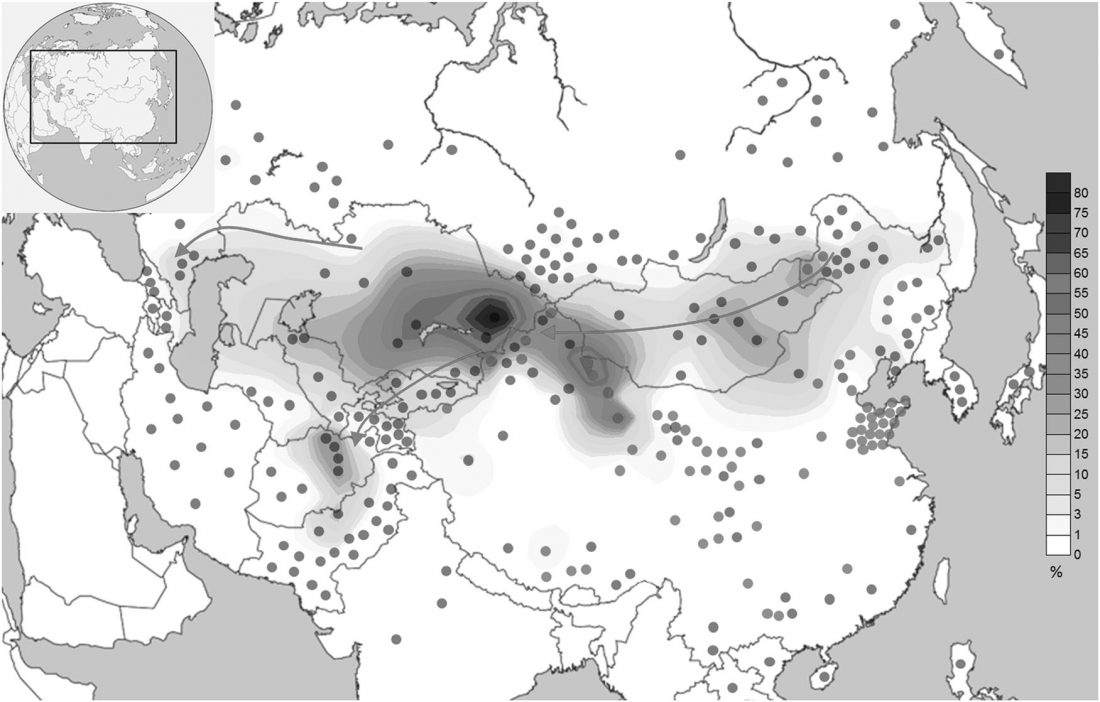
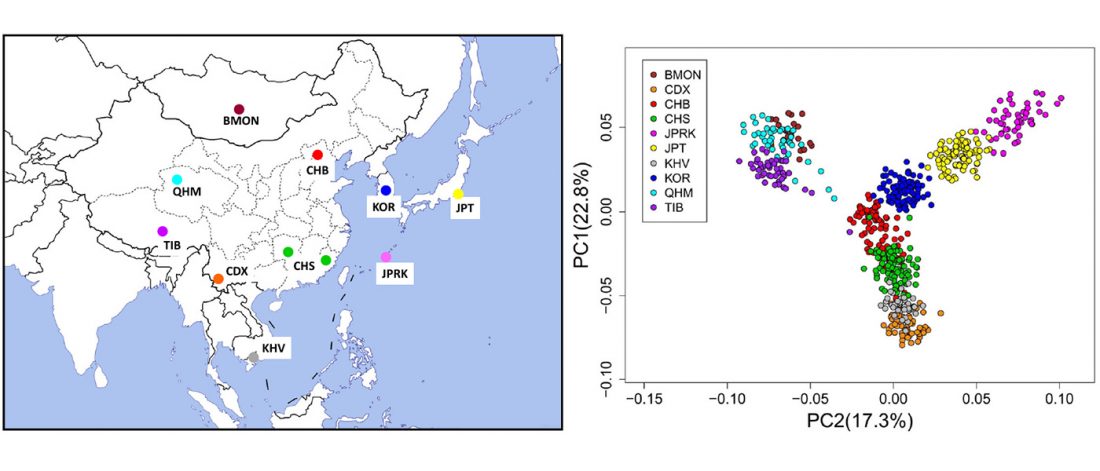
Han Chinese, Japanese and Korean, the three major ethnic groups of East Asia, share many similarities in appearance, language and culture etc., but their genetic relationships, divergence times and subsequent genetic exchanges have not been well studied.Results
We conducted a genome-wide study and evaluated the population structure of 182 Han Chinese, 90 Japanese and 100 Korean individuals, together with the data of 630 individuals representing 8 populations wordwide. Our analyses revealed