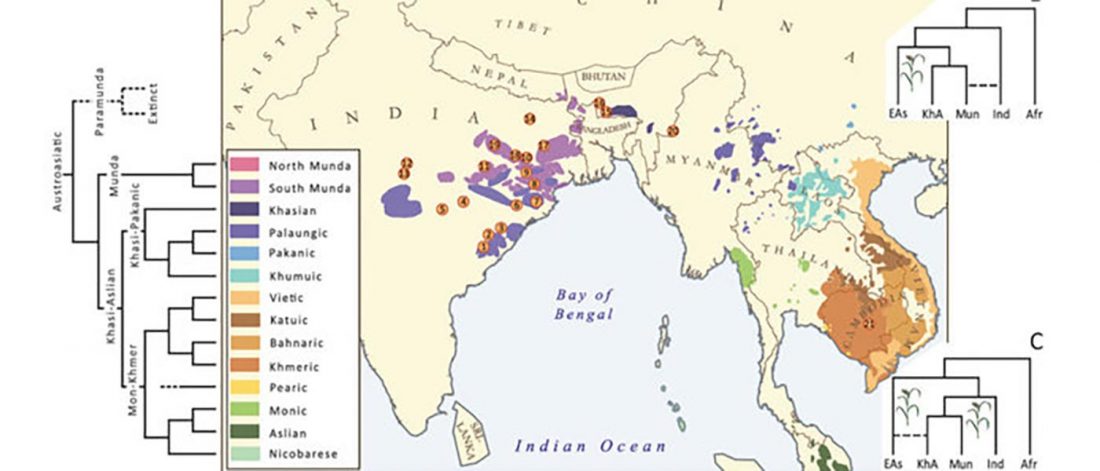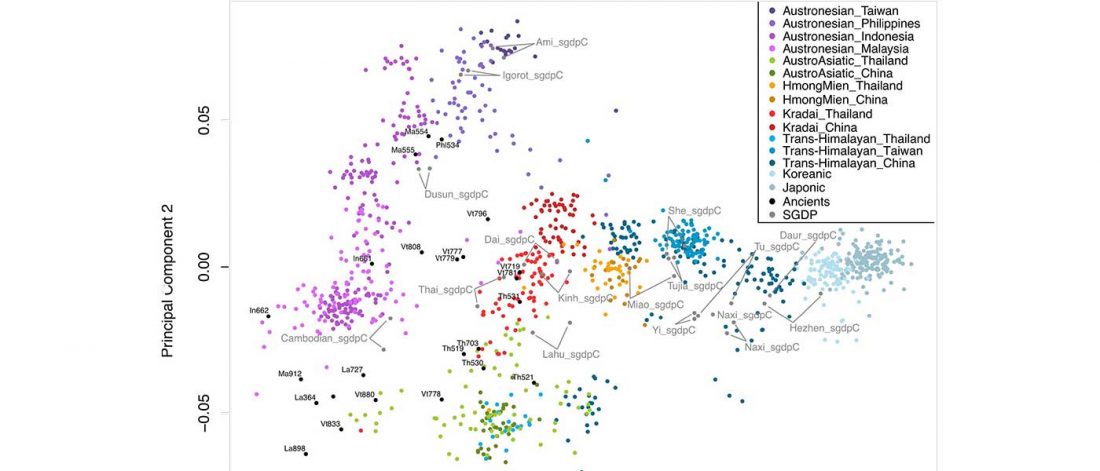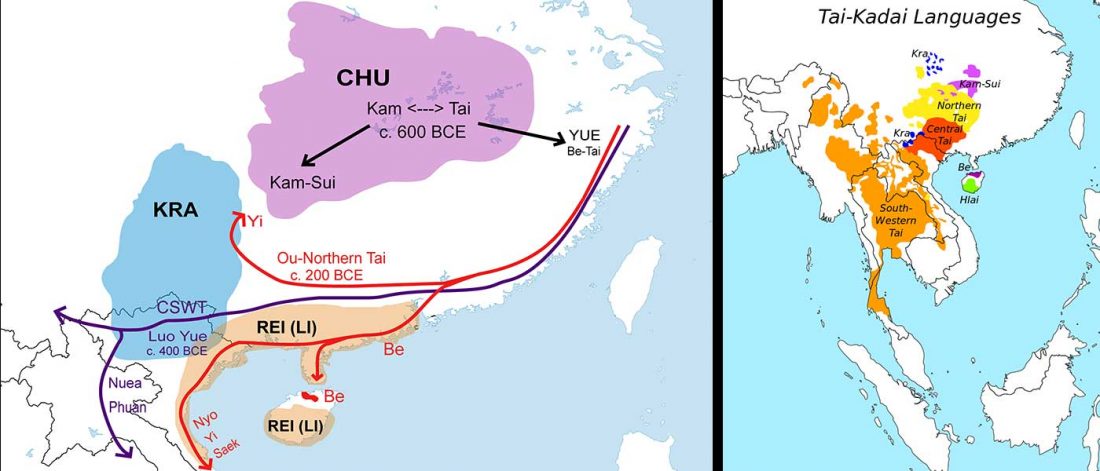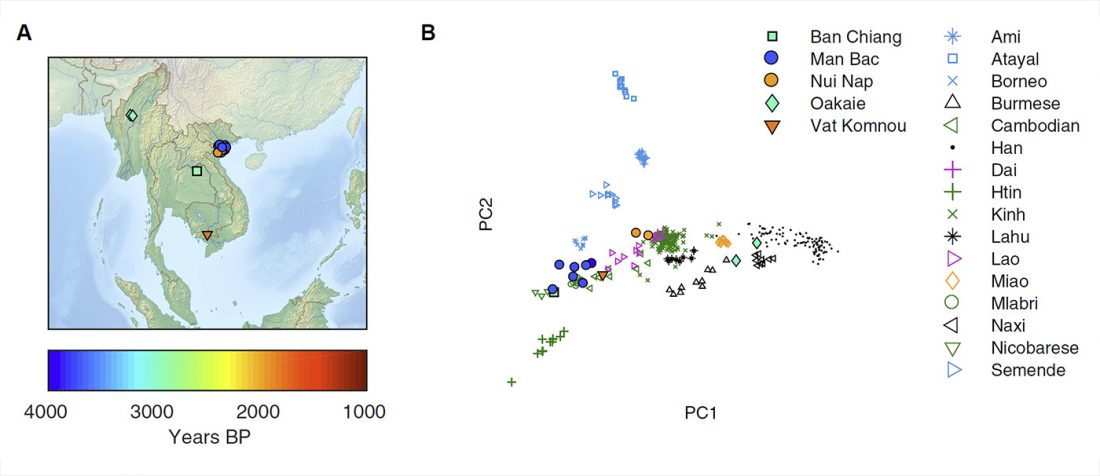
Review (behind paywall) The genetic makings of South Asia, by Metspalu, Monda, and Chaubey, Current Opinion in Genetics & Development (2018) 53:128-133.
Interesting excerpts (emphasis mine):
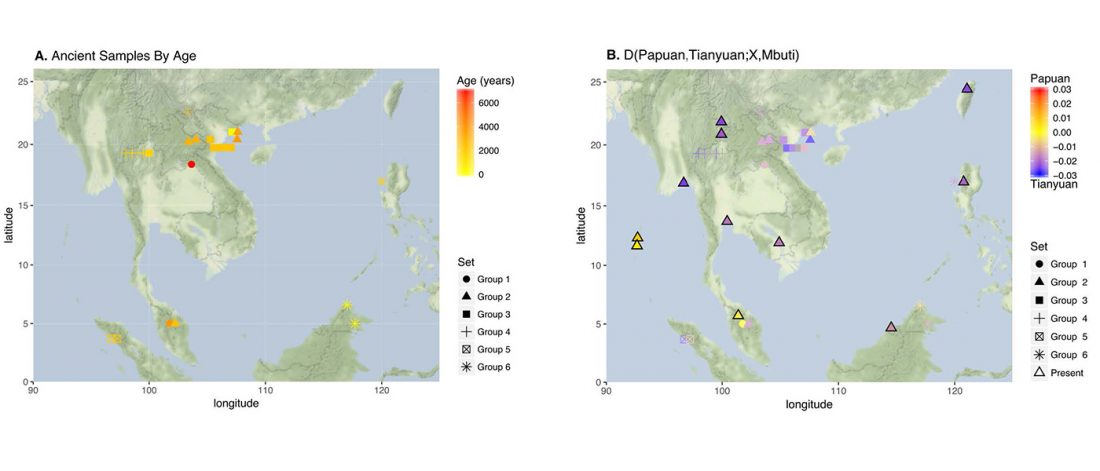
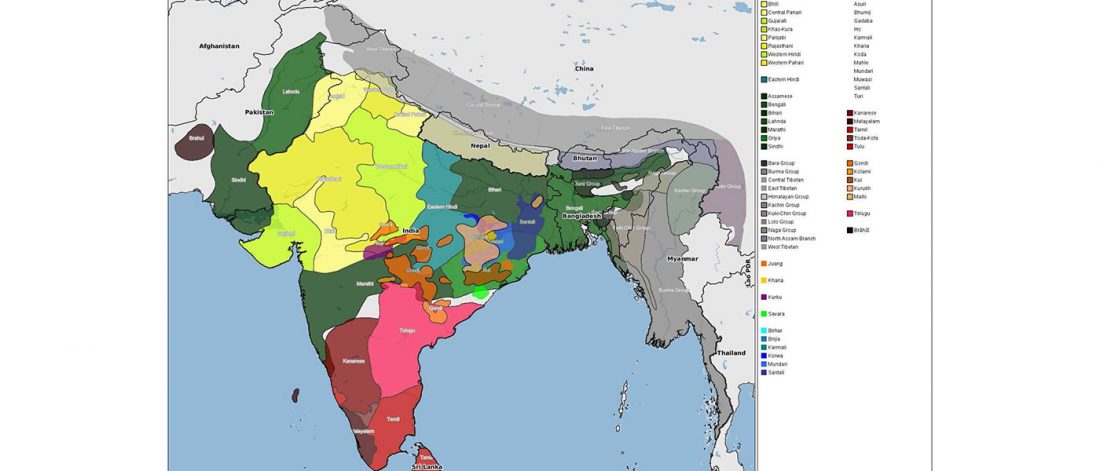
… Read the rest “The genetic makings of South Asia – IVC as Proto-Dravidian”(…) the spread of agriculture in Europe was a result of the demic diffusion of early Anatolian farmers, it was discovered that the spread of agriculture to South Asia was mediated by a genetically completely different farmer population in the Zagros mountains in contemporary Iran (IF). The ANI-ASI cline itself was interpreted as a mixture of three components genetically related to Iranian agriculturalists, Onge and Early and Middle Bronze Age Steppe populations (Steppe_EMBA).