Open access New insights from Thailand into the maternal genetic history of Mainland Southeast Asia, by Kutanan et al. Eur. J. Hum. Genet. (2018) 26:898–911
Abstract (emphasis mine):
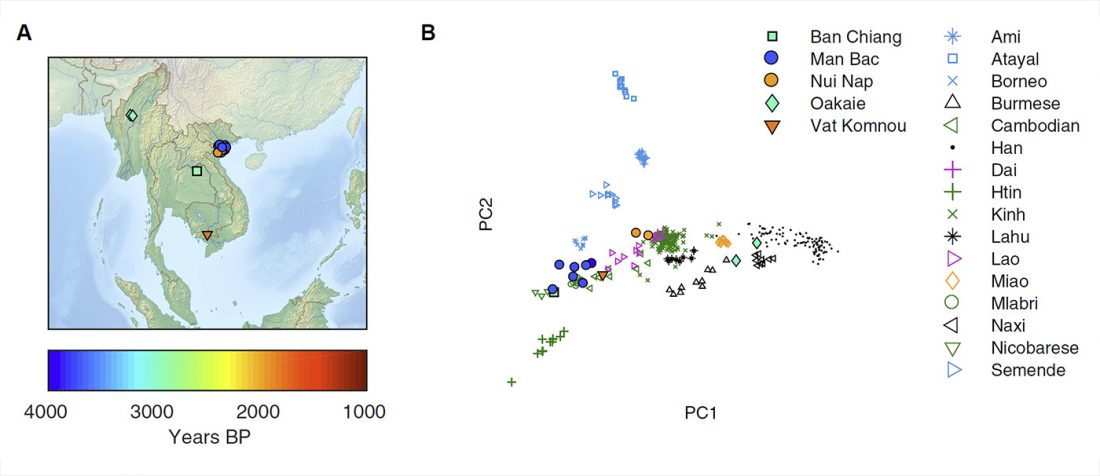
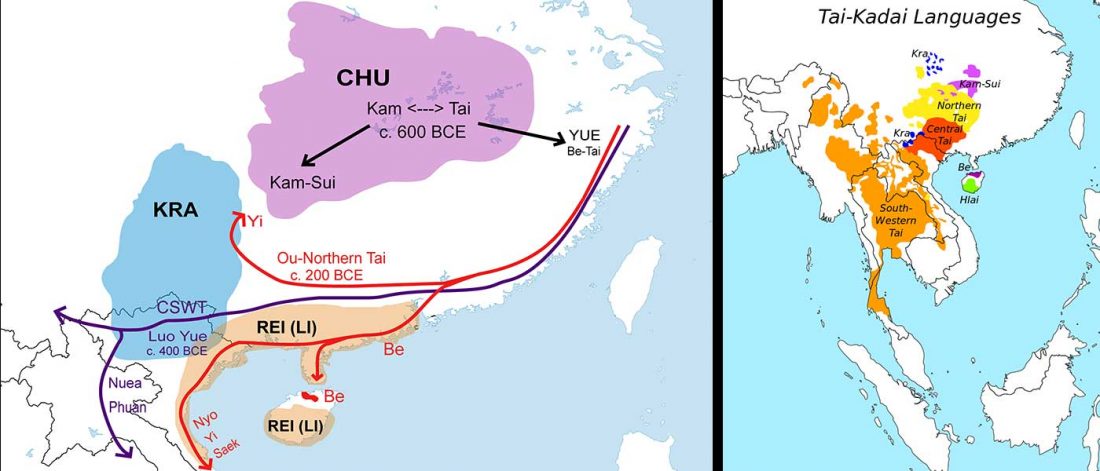
… Read the rest “Mitogenomes from Thailand offer insights into maternal genetic history of mainland South-East Asia”Tai-Kadai (TK) is one of the major language families in Mainland Southeast Asia (MSEA), with a concentration in the area of Thailand and Laos. Our previous study of 1234 mtDNA genome sequences supported a demic diffusion scenario in the spread of TK languages from southern China to Laos as well as northern and northeastern Thailand. Here we add an additional 560 mtDNA genomes from 22 groups, with a focus on the