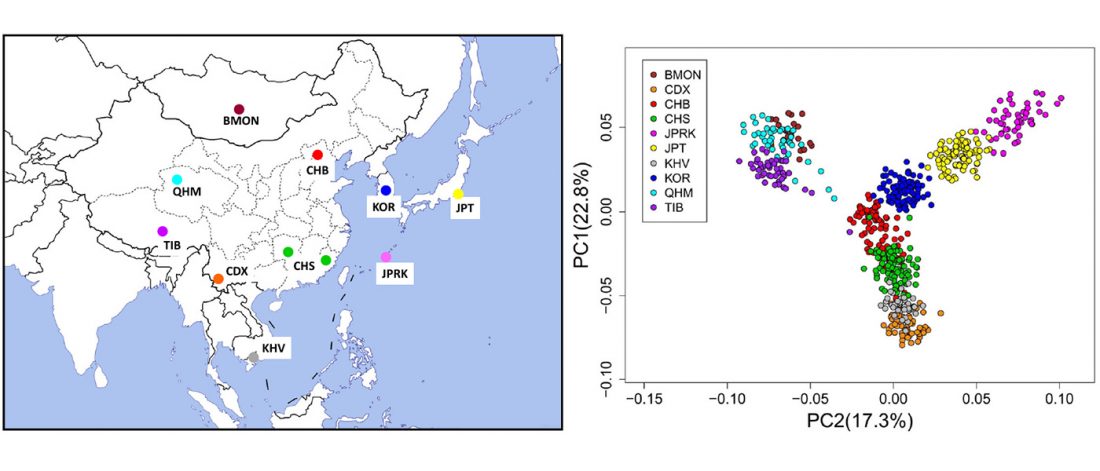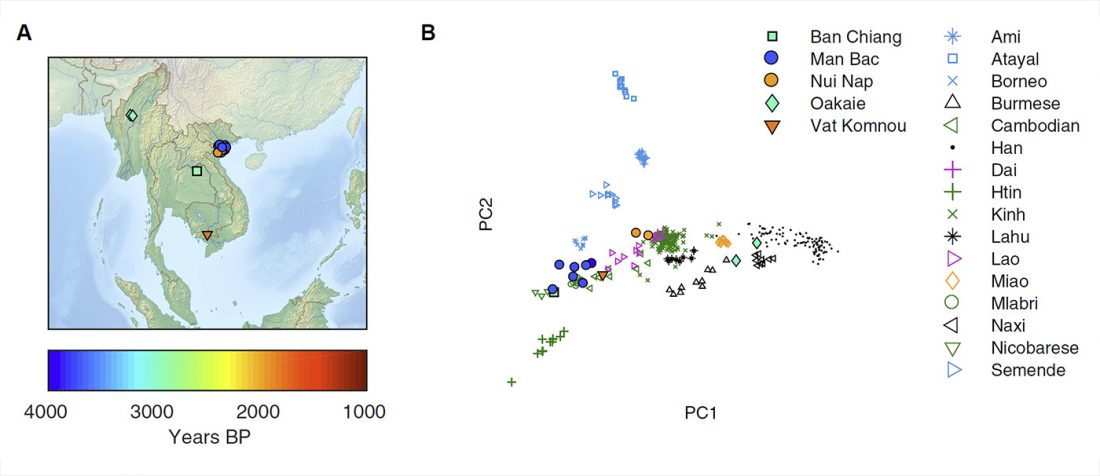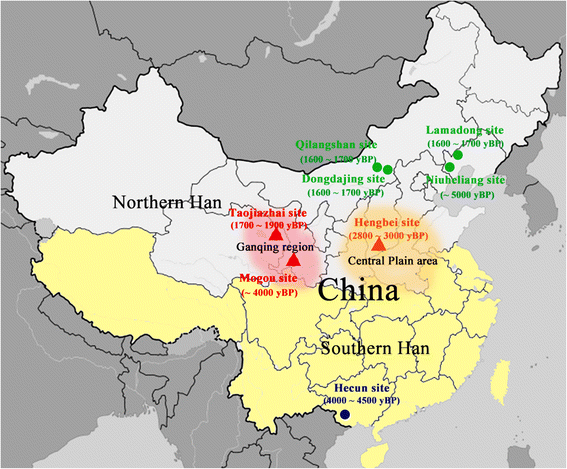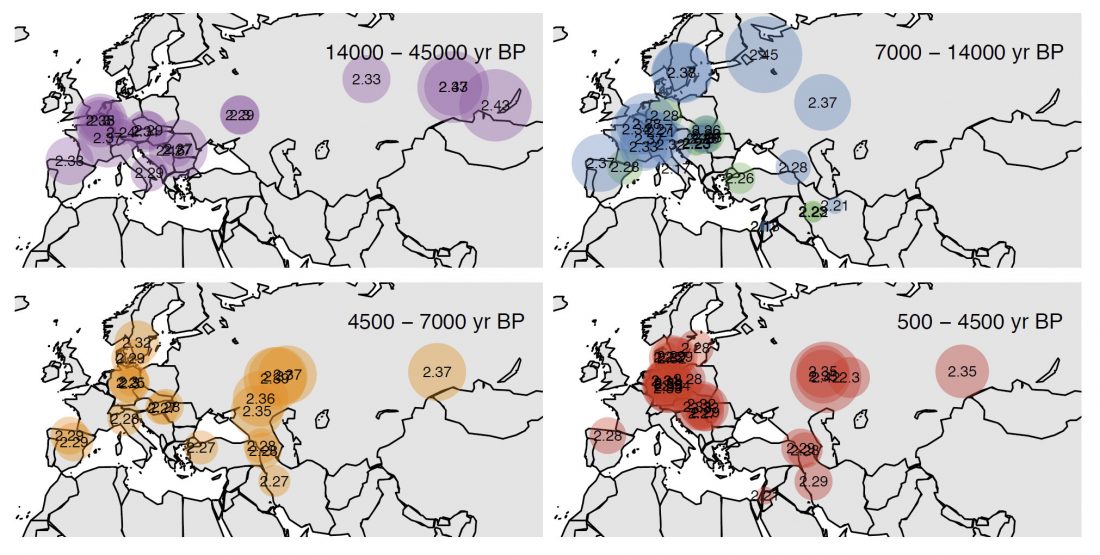
New paper (behind paywall) Reconstruction of Y-chromosome phylogeny reveals two neolithic expansions of Tibeto-Burman populations by Wang et al. Mol Genet Genomics (2018).
Interesting excerpts:
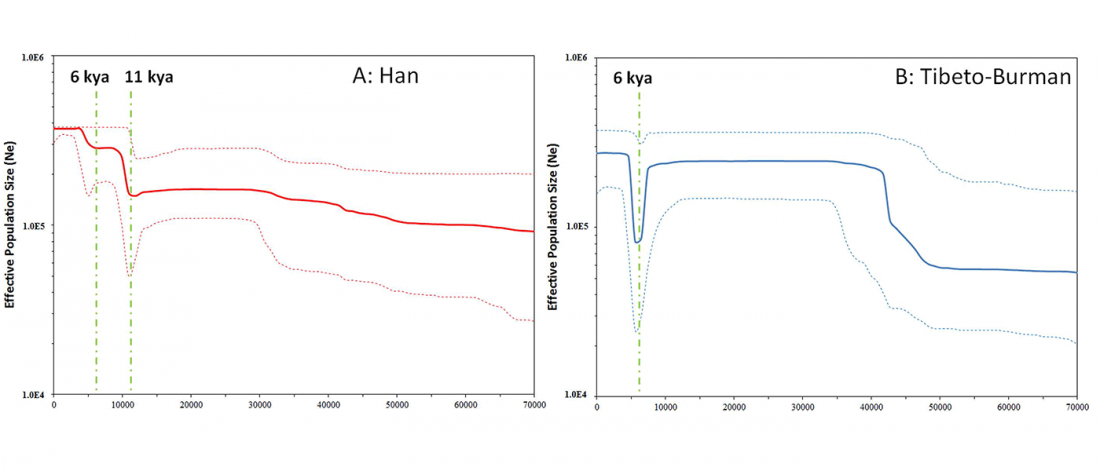
… Read the rest “Reconstruction of Y-DNA phylogeny helps also reconstruct Tibeto-Burman expansion”Archeological studies suggest that a subgroup of ancient populations of the Miaodigou culture (~ 6300–5500 BP) moved westward to the upper stream region of the Yellow River and created the Majiayao culture (~ 5400–4900 BP) (Liu et al. 2010), which was proposed to be the remains of direct ancestors of Tibeto-Burman populations (Sagart 2008). On the other hand, Han populations, the other major descendant group of the Yang-Shao culture (~ 7000–5500 BP), are composed of