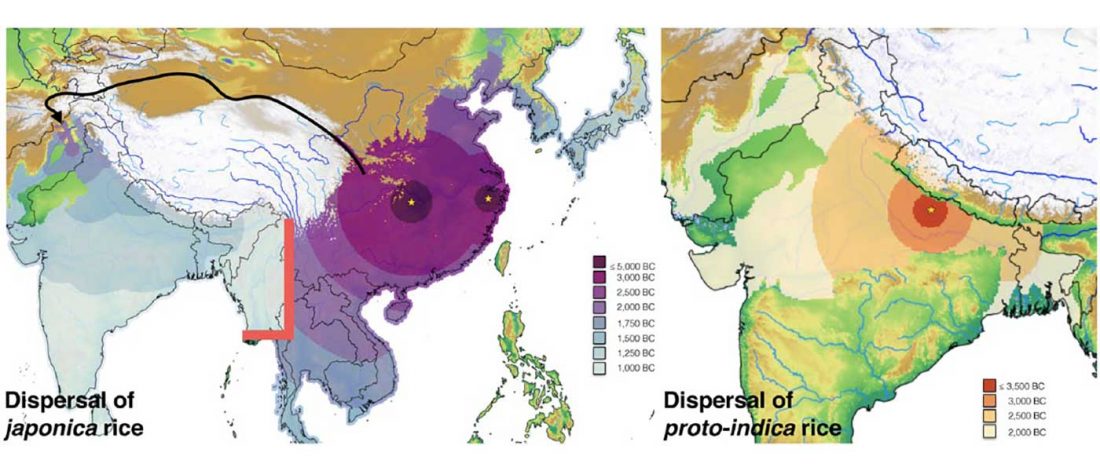
New paper (behind paywall), A tale of two rice varieties: Modelling the prehistoric dispersals of japonica and proto-indica rices, by Silva et al., The Holocene (2018).
Interesting excerpts (emphasis mine):
Materials
… Read the rest “Modelling of prehistoric dispersal of rice varieties in India point to a north-western origin”Our empirical evidence comes from the Rice Archaeological Database (RAD). The first version of this database was used for a synthesis of rice dispersal by Fuller et al. (2010), a slightly expanded dataset (version 1.1) was used to model the dispersal of rice, land area under wet rice cultivation and associated methane emissions from 5000–1000 BP (Fuller et al., 2011). The present dataset (version 2) was used in