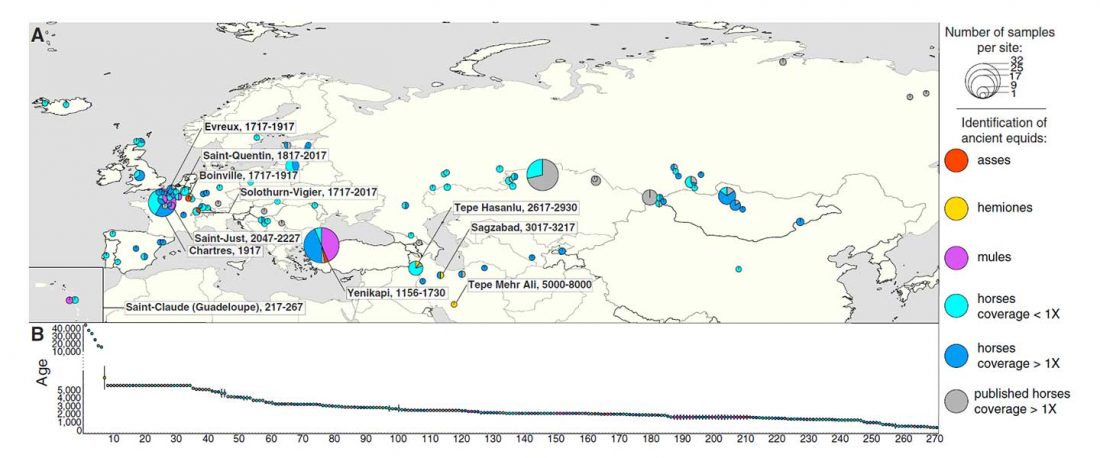
Open access Tracking Five Millennia of Horse Management with Extensive Ancient Genome Time Series, by Fages et al. Cell (2019).
Interesting excerpts (emphasis mine):
… Read the rest “Yamna the likely source of modern horse domesticates; the closest lineage, from East Bell Beakers”The earliest archaeological evidence of horse milking, harnessing, and corralling is found in the ∼5,500-year-old Botai culture of Central Asian steppes (Gaunitz et al., 2018, Outram et al., 2009; see Kosintsev and Kuznetsov, 2013 for discussion). Botai-like horses are, however, not the direct ancestors of modern domesticates but of Przewalski’s horses (Gaunitz et al., 2018). The genetic origin of modern domesticates thus remains contentious, with suggested candidates in the Pontic-Caspian steppes (Anthony, 2007), Anatolia (Arbuckle,