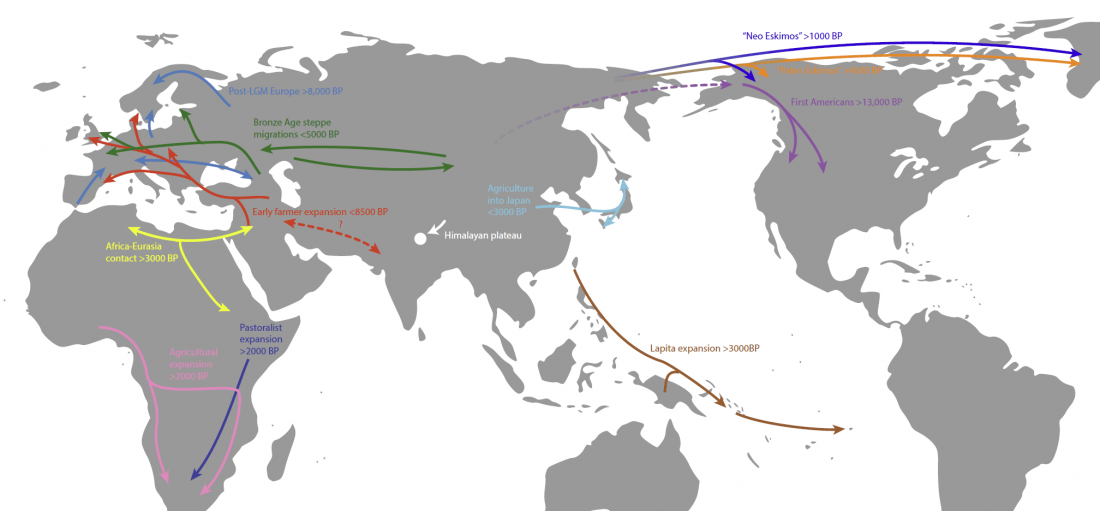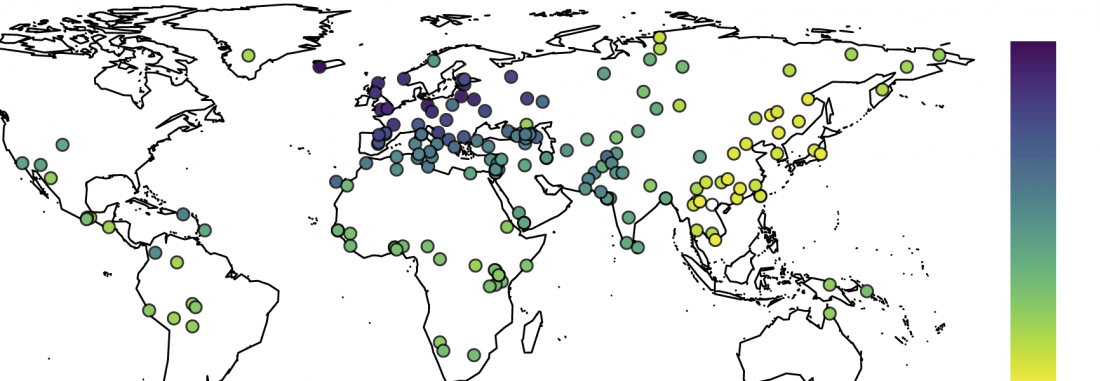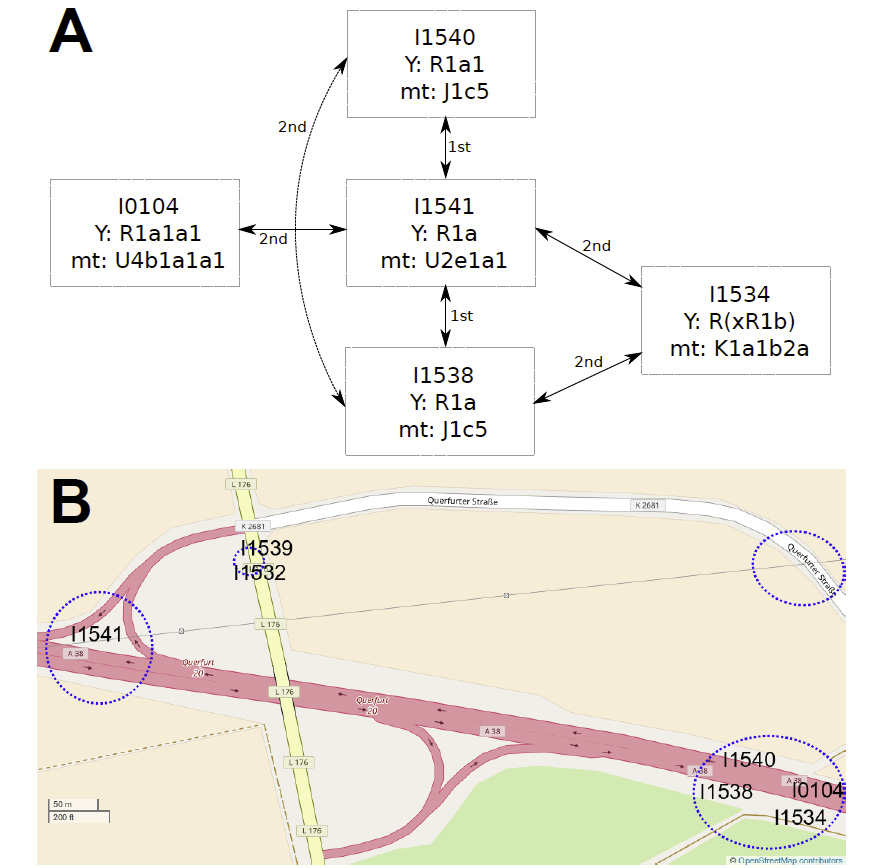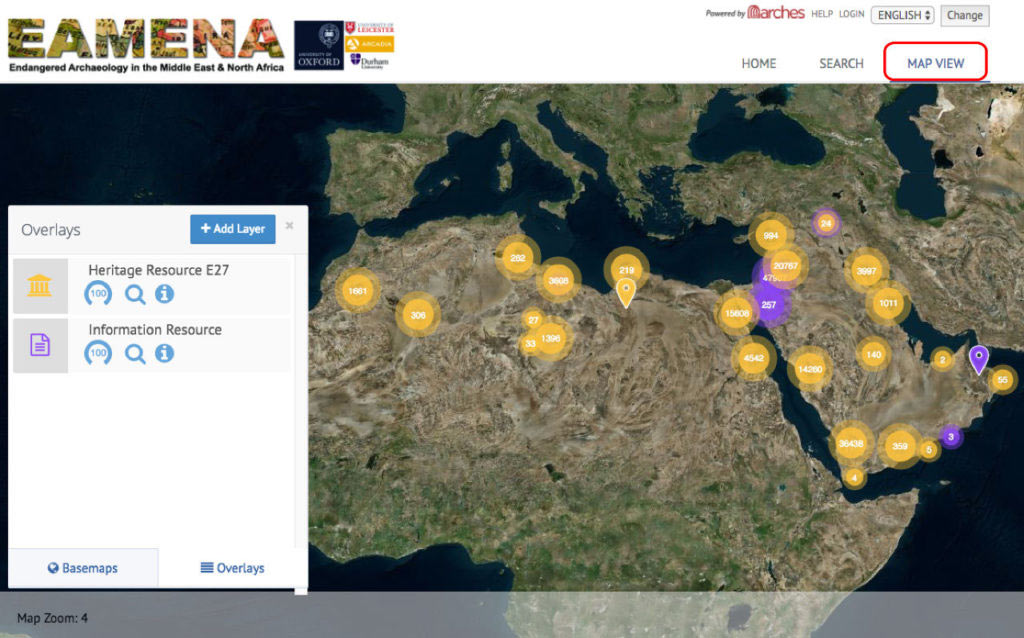
New article, Successful reconstruction of whole mitochondrial genomes from ancient Central America and Mexico, by Morales-Arce et al., Scientific Reports (2017).
Abstract:
… Read the rest “Ancient mtDNA from Central America and Mexico”The northern and southern peripheries of ancient Mesoamerica are poorly understood. There has been speculation over whether borderland cultures such as Greater Nicoya and Casas Grandes represent Mesoamerican outposts in the Isthmo-Colombian area and the Greater Southwest, respectively. Poor ancient DNA preservation in these regions challenged previous attempts to resolve these questions using conventional genetic techniques. We apply advanced in-solution mitogenome capture and high-throughput sequencing to fourteen dental samples obtained from the Greater Nicoya sites of Jícaro